
文摘gydF4y2Ba
基线微生物的特性和功能多样性的人类微生物组使得研究microbiome-related疾病、多样性、生物地理学、和分子功能。美国国立卫生研究院人类微生物组计划提供了一个广泛的这种特征。这里我们介绍第二波的数据研究,包括1631名新基因组(2355)针对不同身体网站有多个时间点在265人。我们应用更新配置和组装方法提供新的微生物个性化的性格特征。应变识别显示亚种演化支特定身体网站;它还量化在隔离基因组物种系统发育多样性并不活跃。全身功能分析途径分为通用、human-enriched和身体site-enriched子集。最后,时序分析分解微生物迅速变异成变量,适度的变量,和稳定的子集。本研究将促进我们的知识基线人类微生物多样性,使个性化的理解微生物功能和动态。gydF4y2Ba
主要gydF4y2Ba
人类微生物组的维护健康不可或缺的组成部分gydF4y2Ba1gydF4y2Ba,gydF4y2Ba2gydF4y2Ba和免疫系统gydF4y2Ba3gydF4y2Ba,gydF4y2Ba4gydF4y2Ba。人口规模的研究已经帮助理解功能的显著后果inter-individual多样性,包括MetaHIT的最早gydF4y2Ba5gydF4y2Ba,gydF4y2Ba6gydF4y2Ba和人类微生物组的项目gydF4y2Ba1gydF4y2Ba(把这里称为HMP1)。研究继续关注肠道gydF4y2Ba7gydF4y2Ba,gydF4y2Ba8gydF4y2Ba,gydF4y2Ba9gydF4y2Ba用更少的人口规模的人群,调查阴道gydF4y2Ba10gydF4y2Ba、口腔gydF4y2Ba11gydF4y2Ba或皮肤gydF4y2Ba12gydF4y2Ba微生物群落。HMP1仍然是最大的全身健康的微生物的扩增子和metagenome调查相结合。gydF4y2Ba
在这里,我们报告的扩展数据集HMP (HMP1-II)组成的whole-metagenome测序(WMS)的1631个新样本HMP队列gydF4y2Ba13gydF4y2Ba(总共2355;gydF4y2Ba扩展数据图1gydF4y2Ba;gydF4y2Ba扩展数据如表1gydF4y2Ba;gydF4y2Ba补充表1gydF4y2Ba)。新样品的数量大大增加科目排序第二和第三,身体和主要目标6网站(总抽样从18岁):前鼻孔,颊粘膜,supragingival斑块,舌背,凳子,后穹窿。质量控制(方法)后,2103年的数据集是由独特的基因组和252技术复制,用于所有以下分析。资料、原始数据和总成公开gydF4y2Bahttp://hmpdacc.orggydF4y2Ba(gydF4y2Ba扩展数据表1 bgydF4y2Ba),gydF4y2Bahttps://aws.amazon.com/datasets/human-microbiome-project/gydF4y2Ba。gydF4y2Ba
全身紧张和生态多样性gydF4y2Ba
菌株的多样性和时空分布首次使用StrainPhlAn调查gydF4y2Ba14gydF4y2Ba(gydF4y2Ba图1gydF4y2Ba),它标识了占主导地位的单体型(应变)的足够丰富的物种在metagenome(方法,gydF4y2Ba补充表2gydF4y2Ba)。大多数先前的文化无关紧张调查只针对肠道gydF4y2Ba15gydF4y2Ba,gydF4y2Ba16gydF4y2Ba和全身系统的距离(距离量化使用木村两个参数gydF4y2Ba17gydF4y2Ba)表明,所有其他栖息地多样性具有更大的应变(gydF4y2Ba图1一个gydF4y2Ba)。与先前的观察一致gydF4y2Ba15gydF4y2Ba,gydF4y2Ba18gydF4y2Ba、应变资料稳定一段时间后,与人们之间的差异随着时间的推移持续低于差异(gydF4y2Ba图1 a, bgydF4y2Ba)。然而,技术差异更低,表明一个基线水平的个体内部应变随时间变化(gydF4y2Ba扩展数据图2 bgydF4y2Ba)。gydF4y2Ba

一个gydF4y2Ba,意味着系统差异gydF4y2Ba17gydF4y2Ba菌株之间的物种有足够的身体覆盖在每一个有针对性的网站(最低2菌株对)。gydF4y2BabgydF4y2Ba,个人倾向于保留个性化的菌株,可视化的主坐标分析(PCoA)的阴谋gydF4y2Ba放线菌gydF4y2Basp.口服分类单元448,线路连接同一个人样本。gydF4y2BacgydF4y2Ba利基协会、量化方法;只有物种有足够的覆盖率至少5个样品在两个或两个以上的身体网站)。值越大表示更大的系统分离机构网站。gydF4y2BadgydF4y2Ba,PCoA利基协会gydF4y2Ba嗜血杆菌56gydF4y2Ba,显示亚种专业化三个不同的网站。gydF4y2BaegydF4y2Ba,PCoAgydF4y2Ba真细菌siraeumgydF4y2Ba。gydF4y2BafgydF4y2Bahuman-associated菌株的报道,目前的16903组参考基因组(方法)。排名前25位的物种的意思是相对丰度当礼物(相对丰度> 0.1%)(最低的50个样本)所示。样本数量gydF4y2Ba补充表2gydF4y2Ba,距离矩阵可从gydF4y2Ba扩展数据表1 bgydF4y2Ba。gydF4y2Ba
几个物种表现出分化成身体特有亚种演化支(gydF4y2Ba图1 cgydF4y2Ba;gydF4y2Ba扩展数据图2得到eugydF4y2Ba),这里定义为离散系统相关的菌株,根据silhouette-based得分利基协会(方法)。这是随时可见的在极端情况下,如gydF4y2Ba嗜血杆菌56gydF4y2Ba(gydF4y2Ba图1 dgydF4y2Ba),不同的亚种中明显的演化支supragingival斑块,颊黏膜、舌背。包括其他物种与显著的特有亚种演化支gydF4y2Ba罗氏菌属mucilaginosagydF4y2Ba,gydF4y2Ba奈瑟氏菌属刺蛾gydF4y2Ba和一个gydF4y2Ba丙酸菌属gydF4y2Ba物种。身体内某些物种没有sub-speciate网站,而是专业演化支不同的个体(例如,gydF4y2Ba真细菌siraeumgydF4y2Ba(gydF4y2Ba图1 egydF4y2Ba),或gydF4y2Ba放线菌johnsoniigydF4y2Ba(gydF4y2Ba扩展数据图2 dgydF4y2Ba));其他人没有离散亚种在人口系统结构(例如,gydF4y2Ba链球菌肝病杂志gydF4y2Ba,gydF4y2Ba扩展数据图2 ugydF4y2Ba)。有趣的是,没有发现特殊亚种演化支,两个城市的研究(gydF4y2Ba扩展数据图2gydF4y2Ba),尽管地理上局部亚种人口结构一直在观察组和更大的地理范围gydF4y2Ba15gydF4y2Ba。gydF4y2Ba
文化无关应变分析,结合16903年NCBI隔离基因组作为参考分析gydF4y2Ba19gydF4y2Ba,提供了一种新的量化gydF4y2Ba20.gydF4y2Ba如何覆盖人类微生物多样性是通过这些引用(gydF4y2Ba图1 fgydF4y2Ba)。Well-sequenced物种如gydF4y2Ba大肠杆菌gydF4y2Ba(gydF4y2Ba扩展数据图2摄氏度gydF4y2Ba)和乳酸杆菌显示小散度从参考隔离。然而,许多普遍和丰富的物种在全身微生物从最近的可用的参考基因组分化明显。明显的演化支缺乏孤立的微生物基因组代表包括在内gydF4y2Ba放线菌gydF4y2Ba(gydF4y2Ba图1 bgydF4y2Ba),gydF4y2Ba嗜血杆菌56gydF4y2Ba(gydF4y2Ba图1 dgydF4y2Ba),gydF4y2Ba真细菌rectalegydF4y2Ba和一些gydF4y2Ba链球菌gydF4y2Ba和gydF4y2Ba拟杆菌gydF4y2Ba孤立的物种,这些代表优先级目标。gydF4y2Ba
由于改进方法和参考基因组,新的了解分类分析包括真核生物,病毒,古菌和额外的54细菌物种在这些相对于HMP1基因组数据gydF4y2Ba1gydF4y2Ba。后者包含普遍的细菌等gydF4y2Ba拟杆菌doreigydF4y2Ba,gydF4y2Ba脆弱拟杆菌gydF4y2Ba,gydF4y2BaAlistipes finegoldiigydF4y2Ba,gydF4y2BaAlistipes onderdonkiigydF4y2Ba和非保密的物种gydF4y2BaSubdoligranulumgydF4y2Ba和gydF4y2BaOscillibactergydF4y2Ba。前者包括gydF4y2BaMethanobrevibactergydF4y2Ba,gydF4y2Ba细胞死亡gydF4y2Ba,gydF4y2Ba假丝酵母gydF4y2Ba(gydF4y2Ba扩展数据图1 cgydF4y2Ba),以及几个病毒:gydF4y2Ba丙酸菌属gydF4y2Ba噬菌体在前鼻孔,gydF4y2Ba链球菌gydF4y2Ba噬菌体在口语网站,和一个gydF4y2BaLactococcusgydF4y2Ba针对C2-like病毒在凳子上。指出寻找同现模式与无菌物种(确切概率法,存在/没有阈值0.1%的相对丰度;gydF4y2Ba补充表3gydF4y2Ba),我们发现gydF4y2BaMethanobrevibacter smithiigydF4y2Ba倾向于共现和几个在肠道梭菌属的物种,包括成员gydF4y2Ba瘤胃球菌属gydF4y2Ba,gydF4y2BaCoprococcusgydF4y2Ba,gydF4y2Ba真细菌gydF4y2Ba,gydF4y2BaDoreagydF4y2Ba(错误发现率(罗斯福)小于0.1),巩固以前的观测gydF4y2Ba21gydF4y2Ba与同现模式和一致的产甲烷菌和梭状芽胞杆菌的精益和肥胖者gydF4y2Ba22gydF4y2Ba。著名的gydF4y2Ba链球菌gydF4y2Ba噬菌体,在口腔最丰富的物种,还与众多共现gydF4y2Ba链球菌gydF4y2Ba物种在口语网站,这表明病毒主要存在前噬菌体,正如前面观察到gydF4y2Ba23gydF4y2Ba。gydF4y2Ba
人类微生物组的核心途径gydF4y2Ba
强有力的患病率(coreness)跨niche-related微生物群落的分子功能可以解释为广泛的分类函数的分布(如基本功能的情况下),或特定的浓缩功能类群中栖息的利基(可能因为函数是选择性有利)。我们调查了这些机制在核心人类微生物组的代谢途径通过功能分析所有使用程序HUMAnN2 HMP1-II样本gydF4y2Ba24gydF4y2Ba(gydF4y2Ba图2gydF4y2Ba;gydF4y2Ba扩展数据图3 gydF4y2Ba,gydF4y2Ba补充表4gydF4y2Ba、方法)。我们专注于1087年代表第一个基因组测序从身体每个主题在6有针对性的访问网站。我们认为的途径是“核心”到一个特定的身体网站(利基)如果是自信地检测到超过75%的人具有较强的分类归属和分类范围符合人类微生物组。从857年开始的一组从MetaCyc量化途径gydF4y2Ba25gydF4y2Ba数据库,我们发现950实例路径的核心机构网站:258通路至少1身体的核心网站,176身体从多个网站的核心地区,和28的核心都是6有针对性的身体网站(gydF4y2Ba图2一个gydF4y2Ba;gydF4y2Ba扩展数据图3gydF4y2Ba)。为了方便,我们将这些类为核心途径,多核途径,分别和supercore通路。gydF4y2Ba

一个gydF4y2Ba总共28身体代谢途径在6大核心网站(“supercore”途径)。一个前鼻孔;Bm,颊粘膜;Pf,后穹窿;年代,凳子;Sp, supragingival斑块;Td,舌背。两个supercore途径和gydF4y2BabgydF4y2Ba,17个额外的途径是在多个身体核心区域和丰富human-associated类群(“人类microbiome-enriched”途径)。gydF4y2BacgydF4y2Ba21通路在1身体更丰富的网站比网站从身体其他地区(“身体site-enriched”途径)。热图值反映了相对丰富的第一四分位数(热图是扩大gydF4y2Ba扩展数据图3 gydF4y2Ba)。在通道栏情节,总(社区)富足是对数,顶部的贡献7个属中按比例缩放。“其他”包含额外的贡献,属;“未分类”包含未知分类的贡献。gydF4y2Ba
区分coreness造成广泛的分类分布与niche-specific浓缩,我们根据其分类范围分类途径(量化的分数non-human-associated属他们在BioCyc注释数据库集合)。虽然大多数的途径进行注释的不到10%的属,核心通路被注释属的34%,多核通路至48%,supercore通路至70%(中间值;所有充实在背景gydF4y2BaPgydF4y2Ba< 0.001,Wilcoxon rank-sum测试)。因此,coreness人体网站通常与广泛的分类分布有关,和通路核心身体更多的网站往往是更广泛的分布式(枪兵gydF4y2BargydF4y2Ba= 0.40;gydF4y2BaPgydF4y2Ba< 0.001;gydF4y2Ba扩展数据图3 bgydF4y2Ba)。极端的例子包括生物合成辅酶A生物合成(见gydF4y2Ba图2一个gydF4y2Ba)和腺苷的核苷酸(gydF4y2Ba扩展数据图3 egydF4y2Ba)两个“管家”功能,不仅广泛分布在人类微生物组,而且所有微生物gydF4y2Ba26gydF4y2Ba,gydF4y2Ba27gydF4y2Ba。虽然我们缺乏对整个MetaCyc通路可分配信息,我们发现单独至关重要的基因家族是更为普遍比非家庭在这些样本(中位数0.94和0.24;Wilcoxon rank-sum测试,gydF4y2BaPgydF4y2Ba< 0.001;方法),符合基本功能被许多机构网站的核心。gydF4y2Ba
相反,19的176多核通路(包括2 supercore通路)自信不是广泛分布,定义保守是不到10%的注释在BioCyc non-human-associated属,和重建的不到10%的pangenomes HUMAnN2数据库(gydF4y2Ba扩展数据图3gydF4y2Ba,gydF4y2Ba4摄氏度gydF4y2Ba)。在这些情况下,人体coreness多个领域被浓缩在human-associated类群中,更好的解释,可能暗示功能适应人类宿主的更广泛的利基。值得注意的是,这些19通路,13(68%)超过两个丰富human-associated属比在BioCyc non-human-associated属,虽然这不是要求他们的定义。人类microbiome-enriched通路包括维生素BgydF4y2Ba12gydF4y2Ba生物合成(从cobinamide adenosylcobalamin救助),这一过程通常执行的微生物群,必须在无菌鼠补充(gydF4y2Ba图2 bgydF4y2Ba)。维生素BgydF4y2Ba12gydF4y2Ba在口腔生物合成也是核心,唾haptocorrin可以保护后在小肠吸收gydF4y2Ba28gydF4y2Ba。丙酸发酵(短链脂肪酸)也特别丰富的口腔和肠道环境(gydF4y2Ba扩展数据图3 fgydF4y2Ba)。短链脂肪酸对他们提出值得注意的作用,维护肠道健康gydF4y2Ba29日gydF4y2Ba,而他们在口腔中的作用研究较少。gydF4y2Ba
最后,个人身体的核心途径特别丰富网站。我们发现一个site-enriched核心途径前鼻孔,七区口腔机构(值得注意的是,几乎没有被浓缩为一个口服网站),从凳子上十个,从后穹窿三(gydF4y2Ba扩展数据图3 dgydF4y2Ba)。硝酸site-enriched通路的例子包括减少口腔(已知的口腔微生物过程与唾液中硝酸盐积累有关gydF4y2Ba30.gydF4y2Ba;gydF4y2Ba图2 cgydF4y2Ba)和甘露聚糖降解肠道(甘露聚糖是人类饮食中的一种植物多糖gydF4y2Ba31日gydF4y2Ba;gydF4y2Ba扩展数据图3 ggydF4y2Ba)。这种site-enriched途径暗示功能适应的微生物群在人体特定的利基。因此,而许多人类微生物组的核心功能反映广泛分布,在全球重要的代谢过程,其他人则可能表明微生物群落适应身体特定网站或人类宿主。gydF4y2Ba
描述颞可变性gydF4y2Ba
新的可用性全身WMS样本在多个时间点每个允许我们进一步描述微生物群落的动态组合在物种水平(gydF4y2Ba图3gydF4y2Ba)。全社区的物种保留利率与以前的观测身体网站除了后穹窿gydF4y2Ba32gydF4y2Ba,gydF4y2Ba33gydF4y2Ba(gydF4y2Ba图3gydF4y2Ba)。个别物种的动力学特征,我们开发了一个高斯过程模型(方法),分解变化丰富的为四部分:本构不同主题,时变动力学(变化可测量的几个月的规模),生物噪声(真出现瞬时变化相对于我们的抽样),噪音和技术(技术之间的复制)。gydF4y2Ba

一个gydF4y2Ba之间,Jaccard相似性最大技术复制和降低随着时间的推移,尽管试相似性总是超过了主客体之间的相似性。gydF4y2BabgydF4y2Ba,方差的高斯过程分解的物种丰度(每个点是一个物种;过滤与生物相关标准方法)为三个组件根据他们的特征时间尺度(方法)。技术噪声估计(gydF4y2Ba补充表5gydF4y2Ba),但没有显示出来。物种具有高不确定性推理(s.e.m.三角图> 0.2)都是灰色的推理是偏向于中心图(方法)。标记版本gydF4y2Ba扩展数据图5gydF4y2Ba。gydF4y2BacgydF4y2Ba一样,gydF4y2BabgydF4y2Ba,但丰度的核心途径。gydF4y2BadgydF4y2Ba内的不同位置上,说明时间序列显示动力学三元图(gydF4y2Ba扩展数据图4 d-fgydF4y2Ba真实的例子)。样本数量gydF4y2Ba补充表1gydF4y2Ba。gydF4y2Ba
这一分析表明哪些物种的身体网站不同大多数个体间,暂时或迅速(gydF4y2Ba图3 bgydF4y2Ba,gydF4y2Ba补充表5gydF4y2Ba,gydF4y2Ba扩展数据图4 d-fgydF4y2Ba)。在肠道中,拟杆菌物种,特别是gydF4y2Ba拟杆菌gydF4y2Ba属(gydF4y2Ba扩展数据图5gydF4y2Ba),主要展出inter-individual变异,而厚壁菌门更暂时在个人动态。口腔和皮肤微生物物种丰度,与此同时,表现出更强的时变动力学和生物整体噪声,与先前的稳定和更个性化的、一致的评估gydF4y2Ba18gydF4y2Ba。一个更详细的看(gydF4y2Ba扩展数据图5gydF4y2Ba)表明,一些物种拥有非常相似的动力学当检测到多个身体站点(例如,gydF4y2Ba罗氏菌属dentocariosagydF4y2Ba)。别人,往往那些特有亚种演化支分析以上,拥有不同的身体之间的动态站点(例如,gydF4y2Ba嗜血杆菌56gydF4y2Ba)。在广泛的范围内,这些物种动力学与先前的协议分析整个社区的动力学在同一队列gydF4y2Ba34gydF4y2Ba。gydF4y2Ba
我们重复这个高斯过程分析来描述的动力学途径丰度以上核心通路识别(gydF4y2Ba图3 cgydF4y2Ba,gydF4y2Ba补充表5gydF4y2Ba)。通路丰度在所有身体网站除了后穹窿个性化比编码的分类单元(远离inter-individual顶点),符合假设社区大会主要是由功能细分市场而不是要求特定的生物gydF4y2Ba35gydF4y2Ba,gydF4y2Ba36gydF4y2Ba。时变氨基酸生物合成途径是丰富(gydF4y2BaPgydF4y2Ba= 0.00025;Wilcoxon rank-sum测试),而inter-individual途径丰富的维生素B生物合成(gydF4y2BaPgydF4y2Ba= 0.00062)。相比之下,阴道微生物显示大型个人组件,在物种和通路水平(所有适应途径inter-individual顶点附近),符合变化在阴道微生物在稳定的社区类型gydF4y2Ba37gydF4y2Ba。肠道功能动态相对较慢,可能反映出对长期趋势因素如饮食模式。相反,在口腔网站快速动力学,特别是在颊黏膜,依照浓缩栖息地的快速能量收获和更大的环境风险。gydF4y2Ba
基因家族发现组装gydF4y2Ba
接下来我们试图建立一个扩展基因目录汇编的基础上扩展的基因组。广泛的基准测试的基础上,我们选择一个自定义使用IDBA-UD装配协议gydF4y2Ba38gydF4y2Ba算法(方法)。而725年HMP1组件生成的gydF4y2Ba1gydF4y2Ba,gydF4y2Ba13gydF4y2Ba,这个协议导致平均装配尺寸,改进中间叠连群长度和将军长度(gydF4y2Ba补充表6gydF4y2Ba)。平均metagenome装配尺寸范围从2.9 megabases (Mb)后穹窿为粪便127.6 Mb。帮助发现新的基因和改善整体装配质量,我们创建了额外co-assemblies从同一个人的集合读取采样在同一身体跨多个网站访问。总的来说,406年和240年co-assemblies相结合是由2和3,分别为(gydF4y2Ba补充表6gydF4y2Ba),装配尺寸大于平均86%的单一组件:中位数装配尺寸从84.8 Mb增加到158.4 Mb,和中位数最大重叠群大小在每个装配从152碱基(kb)增加到167 kb (gydF4y2Ba图4 a - cgydF4y2Ba)。基因的发现是使用MetaGeneMark重叠群上执行gydF4y2Ba24gydF4y2Ba序列分析工具(gydF4y2Ba图4 dgydF4y2Ba;gydF4y2Ba补充表7gydF4y2Ba)。在co-assemblies,基因检测的平均数量从118177增加到213741,而吝啬基因长度保持相似的核苷酸(614比610)。功能的作业是用吸引子(方法)基于几个序列搜索,根据特异性和分类。大约有35 - 45%的基因获得特定的功能注释,在域和另一个30%左右收到注释,家庭,或主题级别(gydF4y2Ba扩展数据图6gydF4y2Ba)。在所有情况下,在每一个特异性的基因数量类别增加co-assemblies,虽然比例仍然类似。因此,尽管从co-assemblies基因预测,他们的注释是特定的单一组件。gydF4y2Ba

一个gydF4y2Ba- - - - - -gydF4y2BadgydF4y2Ba,图基箱线图的总装配尺寸,最大长度和平均叠连群,基因计数单和co-assemblies(样本大小gydF4y2Ba补充表1gydF4y2Ba)。gydF4y2BaegydF4y2Ba基因家族,稀疏曲线(开放阅读框(ORF)集群在90%序列相似性)从单一组件生成预测基因的靶向身体网站(点),用幂律(行)。身体HMP1 WMS数据集的大小在每个网站也显示(圆圈)。稀疏轨迹是健壮的变化序列相似性阈值(188年穹窿样本基因家族的数量只有1131796到1271891之间不等相似之处70 - 95%)。色素是在轴标签gydF4y2Ba一个gydF4y2Ba。样本数量gydF4y2Ba补充表1gydF4y2Ba。gydF4y2Ba
不同的数量,得到包含了gydF4y2Ba39gydF4y2Ba域被reference-based和基于分析倾向于强烈关联在同一个样本(枪兵gydF4y2BargydF4y2Ba= 0.92;gydF4y2Ba扩展数据图7 dgydF4y2Ba),这表明这两种方法提供类似的相对排名社区功能的多样性。此外,这两种方法往往co-detect最核心域包含了身体站点(普遍大于75%;gydF4y2Ba扩展数据图7 egydF4y2Ba)。而reference-based概要文件的存在域包含了基于特征蛋白质的注释,可以直接在装配中发现通过对齐,从而获取新颖的序列多样性。事实上,组装倾向于检测(中位数)19%比reference-based域包含了每个样本的方法,相反倾向于检测建立域包含了与更大的敏感性。这种效果是在前鼻孔网站尤其引人注目,在减少微生物测序深度有限的敏感性相对于reference-based分析大会。gydF4y2Ba
外部数据集相比,总冗余基因集群类似MetaHIT在凳子上gydF4y2Ba6gydF4y2Ba(HMP1-II包含7780363个基因簇,MetaHIT 9879896);相对于现有的滋润皮肤网站基因组gydF4y2Ba12gydF4y2Ba,HMP1-II代表增加780%基因簇(170206到1326693)。然而,即使有成千上万的深度测序人类微生物组在这项研究中,微生物基因家族空间尚未饱和的六个身体检查网站(gydF4y2Ba图4 egydF4y2Ba)。gydF4y2Ba
结论gydF4y2Ba
这里我们提供和分析已知最大全身宏基因组的人类微生物组。相关的深,纵向鸟枪测序使得大规模鉴定新的方面的个性化的微生物。新型分析技术gydF4y2Ba14gydF4y2Ba杰出的暂时稳定的几个物种亚种的人口结构,一些独特的个人和他人身体与特定网站。物种与人类微生物菌株基因组多样性未完全隔离,为隔离和优先次序。新分类剖析解决细菌丰度之间的共生模式和一些古细菌,真核生物和病毒。新的功能分析方法gydF4y2Ba24gydF4y2Ba确定路径所需的人体的微生物定殖,区分这些丰富人类的栖息地从这些普遍的微生物。高斯过程模型微生物特征和功能变化随着时间的推移,和社区发现肠道的组成(特别是拟杆菌物种)相比,高度个性化的其他网站。这个例子表明,肠道拟杆菌门和厚壁菌门的平衡可能不是一个个体的肠道微生物组的定义属性;相反,个人携带拟杆菌门中一个“个人平衡”,和一群系统多样化,暂时变壁厚菌门波动在这个核心。gydF4y2Ba
人类微生物组的许多关键属性特征仍有待甚至在健康人群,除了微生物疾病贡献。需要进一步调查以确定亚种结构识别的功能起源和后果。这种结构也必须全面调查人群,包括地理变异,遗传背景,种族,和环境(例如,HMP1-II北美以外的焦点)。值得注意的是,在这项研究的证据表明,即使在这个相对同质的人群广泛的宏基因组抽样,现存的全部微生物基因尚未测序。与此类似,尽管一个更新元数据之间的共变分析和微生物特性(gydF4y2Ba补充说明gydF4y2Ba;gydF4y2Ba扩展数据无花果8gydF4y2Ba和gydF4y2Ba9gydF4y2Ba)显示几个小说协会、微生物的大多数方差不是解释为协变量测量。HMP1-II,例如,没有测量渡越时间gydF4y2Ba8gydF4y2Ba、免疫状态或参与者的详细的饮食和药物的历史,限制我们的能力来评估这些重要因素。最后,我们对微生物群落的动态和响应的理解必须从这里的描述性模型扩大到包括快速急性扰动的影响。为此,研究时间,更多的密集采样时间的课程需要控制扰动的存在,这里使用的三个时间点。合理修复dysbiotic微生物,因此有必要深化我们对个性化的理解微生物在人类健康。gydF4y2Ba
方法gydF4y2Ba
数据报告gydF4y2Ba
没有统计方法被用来预先确定样本量,这里的数据包括来自biospecimens以前收集在项目第一批人类微生物组研究。包括任何治疗或表型组,没有随机化实验或致盲的进行。gydF4y2Ba
HMP1-II样品和宏基因组测序gydF4y2Ba
样本收集、贮存、处理和WMS测序进行HMP1gydF4y2Ba1gydF4y2Ba。细节IRB审查、知情同意、主题排除标准,抽样协议和时间轴可以在以前的出版物gydF4y2Ba1gydF4y2Ba,gydF4y2Ba13gydF4y2Ba,gydF4y2Ba40gydF4y2Ba。这里所有的基因组分析得到的SRA SRA人类DNA切除后使用BMTagger (gydF4y2Ba扩展数据图7gydF4y2Ba)。SRA原生格式读取所有文件被转换为FASTQ进一步分析使用fastq-dump实用工具的SRA SDK工具包gydF4y2Ba19gydF4y2Ba。gydF4y2Ba
质量控制的核苷酸、读取和样本gydF4y2Ba
一个或多个SRA读文件从每个样本连接每读方向来创建一个为每个样本对FASTQ文件。这些FASTQs转化为不结盟的BAM使用皮卡德(gydF4y2Bahttp://broadinstitute.github.io/picard/gydF4y2Ba)和确切的副本被皮卡EstimateLibraryComplexity模块的修改版本。最后,所有的阅读都修剪和长度过滤(q2 -l60)使用trimBWAstyle.usingBam。pl脚本从加州大学戴维斯分校生物信息学核心基因组中心(gydF4y2Bahttps://github.com/genome/genome/blob/master/lib/perl/Genome/Site/TGI/Hmp/HmpSraProcess/trimBWAstyle.usingBam.plgydF4y2Ba)。gydF4y2Ba
分类分析(下图)后,生态异常WMS样品被确认为进一步样品质量控制基于中值了解Bray-Curtis不同身体其他样本相同的网站。如果样本不同的中位数超过了上层内部围栏(四分位范围的1.5倍以上第三个四分位数)值从它的身体站点的异同,样本标签局外人和丢弃。这个过程移除86 (3.6%)WMS样本对各自的身体高度非典型网站。下游分析使用剩余的2355个样本。gydF4y2Ba
分类和应变分析gydF4y2Ba
分类使用MetaPhlAn2宏基因组进行样品的分析gydF4y2Ba20.gydF4y2Ba,它使用一个库clade-specific标记提供panmicrobial(细菌、古细菌、病毒和真核)分析(gydF4y2Bahttp://huttenhower.sph.harvard.edu/metaphlan2gydF4y2Ba)。从HMP1 MetaPhlAn2资料完成观察生态模式(gydF4y2Ba扩展数据图1 bgydF4y2Ba),同意直接读取映射到参考基因组。映射读取覆盖81.7%的平均(平均92.8%)的每个小幅优势毒株的参考基因组序列(包含至少5%的社区)所有样本。意味着覆盖深度(总碱基对排列参考基因组中读取除以总碱基对)对这些菌株对所有样品是3.9×,覆盖的地域广度意味着变化很大,身体网站从0.04×(右肘前的窝)到11.1×(舌背)(gydF4y2Ba补充表8gydF4y2Ba)。批处理效果不可见的两个轴的变化在每个身体网站(gydF4y2Ba扩展数据图1 dgydF4y2Ba)。gydF4y2Ba
使用StrainPhlAn应变特性进行gydF4y2Ba14gydF4y2Ba。StrainPhlAn描述单核苷酸变异MetaPhlAn2标记基因的有机体。对于一个给定的样本,我们需要至少80%的标记对于一个给定的物种有一个最小均10×深度阅读,以确保足够的数据进行单体型调用。总共151种满足这些需求在至少两个WMS样本(gydF4y2Ba补充表2gydF4y2Ba)。之间的距离使用木村两个参数距离菌株进行了评估gydF4y2Ba17gydF4y2Ba(可以从gydF4y2Ba扩展数据表1 bgydF4y2Ba)。MetaPhlAn2和StrainPhlAn都使用默认设置。gydF4y2Ba
参考基因组覆盖率由补的得分不对称系统的距离(1−UniFrac GgydF4y2Ba41gydF4y2BaHMP1-II菌株和参考基因组之间)。在所有报道估计gydF4y2Ba补充表2gydF4y2Ba。gydF4y2Ba
Niche-association得分gydF4y2Ba
物种niche-associated亚种演化支被测量发现类似剪影得分,这比较意味着系统发育差异的菌株在每个机构网站的散度菌株(在同一物种)跨越身体网站。具体来说,我们首先定义一个身体网站不同的分数gydF4y2BaDgydF4y2Ba(gydF4y2BaugydF4y2Ba,gydF4y2BavgydF4y2Ba)对于一个给定的物种在机构网站gydF4y2BaugydF4y2Ba和gydF4y2BavgydF4y2Ba为:gydF4y2Ba
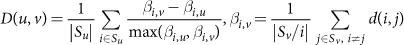
在哪里gydF4y2Ba年代gydF4y2BaxgydF4y2Ba的样本集通过StrainPhlAn覆盖率要求身体的网站gydF4y2BaxgydF4y2Ba,gydF4y2BadgydF4y2Ba(gydF4y2Ba我gydF4y2Ba,gydF4y2BajgydF4y2Ba木村)是两个参数主要单样本之间的距离gydF4y2Ba我gydF4y2Ba和gydF4y2BajgydF4y2Ba。niche-association分数gydF4y2Ba一个gydF4y2Ba为每一个物种(gydF4y2Ba图1 bgydF4y2Ba)被定义为最大gydF4y2BaDgydF4y2Ba(gydF4y2BaugydF4y2Ba,gydF4y2BavgydF4y2Ba)所有导演对身体的网站gydF4y2BaugydF4y2Ba和gydF4y2BavgydF4y2BaStrainPhlAn覆盖需求得到满足至少5个样品在这两个网站。也就是说,一组身体的网站gydF4y2BaBgydF4y2Ba:gydF4y2Ba
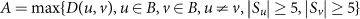
这方面的一个担忧是,更大的技术困难single-nucleotide-variant调用在一个站点可能导致明显的利基协会没有。这不是一个问题在这里,然而,随着网站niche-association分数计算的都是口语网站相似的技术变化(gydF4y2Ba图1一个gydF4y2Ba)。这是一个副产品的限制物种被要求有足够的存在(5样品通过StrainPhlAn覆盖需求)在多个站点,这是不可能的以外的生态更相似的一组口服网站。gydF4y2Ba
功能分析gydF4y2Ba
功能分析是使用HUMAnN2执行gydF4y2Ba24gydF4y2Ba(gydF4y2Bahttp://huttenhower.sph.harvard.edu/humann2gydF4y2Ba)。简而言之,对于一个给定的样本,从pangenomes HUMAnN2构造sample-specific引用数据库子集的物种样本中检测到的MetaPhlAn2 (pangenomes orf的预先计算的表示给定的物种gydF4y2Ba42gydF4y2Ba)。HUMAnN2然后地图样本对该数据库读取量化基因的存在和丰富在每个物种的基础上。剩余未映射读取进一步映射通过翻译搜索UniRef-based蛋白质序列目录gydF4y2Ba43gydF4y2Ba。最后,对于基因家族量化核苷酸和蛋白质含量,HUMAnN2采样通路的功能特征子集和评估社区总species-resolved,非保密途径丰度基于MetaCyc通路数据库gydF4y2Ba44gydF4y2Ba。gydF4y2Ba
分析代谢途径coreness都集中在1087年HMP1-II代表第一个基因组测序从每个主题的六个目标主体访问网站。后续样品和技术复制对于一个给定的(网站主题,正文)被排除在外,以避免偏压组合人口估计的方向。我们定义了一个“核心”路径在一个特定的身体部位,检测相对丰度> 10gydF4y2Ba−4gydF4y2Ba至少在subject-unique样本的75%。我们进一步过滤这些高度流行的途径,以确保合理的分类范围和自信的分类归属。具体地说,一个潜在的核心途径如果BioCyc排除在外gydF4y2Ba44gydF4y2Ba带注释的分类范围不包括任何human-associated微生物属(定义为属中发现至少5 HMP科目相对丰度> 10gydF4y2Ba−3gydF4y2Ba),或者如果> 50%的通路副本“未分类”分类归因> 25%的样本。这些过滤条件产生了950核心(通路、身体站点)协会覆盖258独特的MetaCyc通路。值得注意的是,这些数字是相当不准确的参数设置上面所描述的那样,只要coreness的整体定义包含(1)多数人口患病率(> 50%),(2)论文检测阈值(即低于(途径)gydF4y2Ba1gydF4y2Ba),和(3)某种形式的分类过滤限制假阳性(例如,否则共同通路的罕见变异;gydF4y2Ba补充表9gydF4y2Ba)。gydF4y2Ba
我们量化的分类范围的途径的一部分它独特的属BioCyc注释。我们这个标准细分成范围“human-associated”和“non-human-associated”属(如上面定义),和关注后者措施避免循环论证(一个函数,它是广泛分布在human-associated类群丰富人类微生物组的定义)。进一步控制,我们也直接应用HUMAnN2底层pangenome数据库关联路径> 4000微生物物种。保守定义核心途径丰富人类微生物组”,我们要求他们注释< BioCyc non-human-associated属的10%,还直接注释< 10%的non-human-associated pangenomes。第二准则进一步降低的共同通路的罕见变异(MetaCyc所定义的)被称为基因组由于cross-detection丰富的共同通路。gydF4y2Ba
我们定义了一个核心途径身体强烈浓缩在一个特定的网站如果第一个四分位数的丰富通路在那个网站是> 2×大于第三四分位数从所有其他的身体地区丰富的网站(也就是说,焦点和背景丰度分布必须很好地分离,而不仅仅是明显不同)。值得注意的是,这个定义只需要核心途径口服身体网站独立的从非对话网站作出,而不是其他口服网站(很少的核心途径口服身体强烈丰富网站相对于其他口服网站)。gydF4y2Ba
我们调查coreness之间的关系和重要性函数使用一个数据集的大约300个至关重要的齿轮gydF4y2Ba45gydF4y2Ba基因家族决定在gydF4y2Ba大肠杆菌gydF4y2Ba46gydF4y2Ba(“义塾集合”)。我们计算齿轮丰富基因组在1087年推出了上面总结丰富的个人UniRef基因家族(根据UniProt-derived HUMAnN2计算)齿轮注释gydF4y2Ba47gydF4y2Ba。我们认为一个齿轮是自信的,如果样品中检测出其相对丰度超过10gydF4y2Ba−4gydF4y2Ba。在检测齿轮基本齿轮(gydF4y2BangydF4y2Ba= 272)都是在全球范围内普遍比非齿轮(gydF4y2BangydF4y2Ba= 3629;中位数0.94和0.24)和核心更多身体网站(平均4.7和1.2;核心在这里定义为> 75%流行网站);这两个趋势具有高度统计学意义(gydF4y2BaPgydF4y2Ba< 0.001)由Wilcoxon符号秩检验和健壮的一个较小的检测阈值(10gydF4y2Ba−6gydF4y2Ba)。gydF4y2Ba
高斯过程动力学建模gydF4y2Ba
高斯过程是一个非参数概率模型进行推断取样连续函数。本节讨论具体的理由高斯过程模型用来模拟微生物和功能丰度(把这里称为“特性”)的微生物,并讨论了其假设,优点和缺点。提出了实现细节在以下部分。gydF4y2Ba
高斯过程,模拟的联合分布函数在任何有限点集遵循多元正态分布。不失一般性,高斯过程可以仅仅通过他们的协方差函数或内核参数化,定义输出的协方差之间的任何两个采样点。这个成对定义允许使用不规则的时间采样HMP1-II数据集(中gydF4y2Ba扩展数据图4gydF4y2Ba)。高斯过程的协方差函数的形状决定了建模函数的几个性质,如平滑、速度变化,哪些特性的输入向量是敏感的。因此我们的第一目标是评估几种常见的证据的强度协方差函数描述生物学上有意义的行为,并确定哪些组件应该被包括在一个简洁的模型来捕获大部分的可观测的动力学特性。候选人的协方差函数集我们考虑包括:快速变化(生物噪声),inter-individual差异,一个Ornstein-Uhlenbeck过程,squared-exponential协方差函数,和季节动态与一年(公式中可以找到gydF4y2Ba补充表10gydF4y2Ba)。gydF4y2Ba
所有候选人协方差函数描述静止的过程,考虑到固有的有限状态空间的相对丰度,尽管他们有不同的时间动力学和生物系统产生这些行为的影响。“快速变化”,即在时间尺度上变化的速度比衡量,由高斯白噪声过程。Inter-individual样本之间的差异是由恒协方差为同一个人。两个时变组件,Ornstein-Uhlenbeck过程和squared-exponential协方差函数,两个单调减少协方差描述为两个样品之间的差异时间增加;也就是说,时间点接近另一个比之间的距离会更相似。这两个函数主要是不同的底层函数的光滑性。Ornstein-Uhlenbeck过程是唯一与非平凡的协方差平稳高斯马尔可夫链的过程随着时间的推移,和生产函数,不是可微的,因此非常参差不齐的,类似布朗运动。例如,预计这个协方差函数的丰度缓慢变化特性的连续随机扰动环境下。与此同时,squared-exponential协方差函数描述无限可微的函数,因此非常光滑。这个函数意味着大量的潜伏状态过程中生成大量的相关特性。 Both of these time-varying covariance functions are parameterized by their length scale, the characteristic time scale at which the function changes. Lastly, the seasonal component is represented by the canonical periodic covariance function from Gaussian process literature, with its period fixed at one year, but with an unknown length scale. Here, a model refers to a combination of these covariance functions.
模型基于边际相比可能(也称为“证据”),位(也就是说,日志中报告gydF4y2Ba2gydF4y2Ba边际可能比=日志gydF4y2Ba2gydF4y2Ba贝叶斯因子)的证据对给定模型的最佳模型相比,功能(gydF4y2Ba补充表10gydF4y2Ba)。超过3.3比特对一个模型被认为是强有力的证据,和超过6.6被认为是决定性的。边际可能是从马尔可夫链蒙特卡罗(密度)估计样本的后验分布截断调和平均数未规范化后验分布的采样点。截断了,这个估计是已知可怜的收敛特性,因为密度和样品非常低的可能有不合理影响谐波的意思。比较对模型进行适合十大最普遍的物种的丰度(至少70%的非零丰度)和五大最丰富的通路在每个目标身体网站(gydF4y2Ba补充表10gydF4y2Ba)。比较也进行了一系列的模拟特性与已知动态(“控制”),从相应的采样的高斯过程由于技术噪声方差的5%,剩余方差组件之间均匀分布。gydF4y2Ba
,以确定哪些这些组件的统计数据支持,我们使用一个标准的贪婪搜索可能的空间模型,从最简单的模型开始(所有变异是技术)和迭代拒绝简单的模型支持更复杂的一个,如果证据对简单模型超过六位。更复杂的模型被认为是在每次迭代那些只有一个参数,并包含简单的模型作为一个特例(伪代码gydF4y2Ba补充表6gydF4y2Ba)。这个过程选择的模型,包括两个简单的组件,生物噪声和inter-individual差异,47岁,53 * 72特性测试,分别。在更复杂的组件,Ornstein-Uhlenbeck组件选择的13倍,而squared-exponential协方差函数和季节性组件被选为一个测试功能。这些趋势是健壮的模型中增加拒绝阈值,与其余Ornstein-Uhlenbeck组件重要的证据至少10位,而squared-exponential协方差函数和季节性组件只选择更加宽松的阈值(≤4比特)。然而,我们注意到,这个过程很难识别squared-exponential协方差函数和季节性组件控制样本,包括其他组件(特别是生物噪声),表明这些组件是很难区分的可用时间采样模式。因此,尽管目前数据明显喜欢Ornstein-Uhlenbeck组件squared-exponential协方差函数,和不支持的一个季节性的组件,我们没有足够的动力来消除这些潜在的重要贡献者微生物的动态。最后,零模型只有技术噪声被拒绝了71的73的特性,往往非常高的证据(中位数69.6比特)。gydF4y2Ba
剩余部分的分析,我们因此聚集在一个模型有四个组成部分:inter-individual差异,一个Ornstein-Uhlenbeck过程,生物噪音,噪音和技术。让gydF4y2BaUgydF4y2Ba,gydF4y2BaTgydF4y2Ba,gydF4y2BaBgydF4y2Ba,gydF4y2BaNgydF4y2Ba这些组件各自的大小,gydF4y2BalgydF4y2BaOrnstein-Uhlenbeck过程的时间尺度。估计这些参数(hyperparameters高斯过程命名)是由与下面的协方差函数拟合高斯过程,所有功能(物种和通路)和至少75%的患病率在一个网站(gydF4y2Ba图3gydF4y2Ba,gydF4y2Ba补充表5gydF4y2Ba):gydF4y2Ba
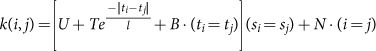
这个函数描述样本i和j之间的协方差,在哪里gydF4y2BatgydF4y2BaxgydF4y2Ba和gydF4y2Ba年代gydF4y2BaxgydF4y2Ba分别为样本的抽样日期和主题标识符gydF4y2BaxgydF4y2Ba。四个参数都符合同时获得(下一节)。自三个级组件必须和人口的变化,这可以视为一个方差分解成不同的可变性来源时间签名。我们只在这里的三个生物组件感兴趣,因此,我们正常的噪音估计技术组件(也就是说,gydF4y2BaUgydF4y2Ba,gydF4y2BaTgydF4y2Ba,gydF4y2BaBgydF4y2Ba]gydF4y2BaNgydF4y2Ba)可视化标准三元分解图(gydF4y2Ba图3 b, cgydF4y2Ba)。插图,我们展示三个例子说明图的三种类型的动力学设计为允许数据和之间的直接比较符合高斯过程(gydF4y2Ba扩展数据图4 d-fgydF4y2Ba)。gydF4y2Ba
时间依赖模型的任何组件的可识别性是有限的时间采样模式可用。当前数据集只包含每人三个时间点,与样品之间的时间大约在一个月和一年之间均匀分布为每个站点(身体gydF4y2Ba扩展数据图4gydF4y2Ba)。过程太快,测量将为生物噪声分量,而过程慢得多比的最长时间间隔可以有助于inter-individual组件。我们测试时间尺度是什么Ornstein-Uhlenbeck检测到的组件,并将有助于inter-individual或生物噪声组件,通过模拟数据从Ornstein-Uhlenbeck流程可变长度的尺度和执行参数符合(gydF4y2Ba扩展数据图4 bgydF4y2Ba)。这些表明,时变组件是敏感的过程与特征长度尺度约3 - 24个月。gydF4y2Ba
我们注意到解决时变组件是唯一可能的,因为大型传播时间HMP1-II中可用的样本数据集之间的差异(gydF4y2Ba扩展数据图4gydF4y2Ba)。另一个常见的纵向研究设计中,少量的样本收集每人有一个固定的时间间隔,这是不可能的,尽管这种设计可能使分析更简单(样品可以按时间点和分组方法如高斯过程不会是必要的)。同样的,更丰富的纵向数据长时间序列的形式将允许更多的推断关于微生物的动态。特别感兴趣的,这将使不同的时间人们之间的组件(s)来解决。在这里,每人只有三个时间点,适合描述时序模型参数的变化(gydF4y2BaBgydF4y2Ba,gydF4y2BaTgydF4y2Ba,gydF4y2BalgydF4y2Ba)只有一个最佳的人口。这样的采样模式也会提供机会来区分更确切之间的马尔可夫链的Ornstein-Uhlenbeck过程和其他可能的non-Markovian过程(如squared-exponential所描述的协方差函数,或一个中间如Matern协方差函数),表明潜在的大迟延事件状态或微生物。gydF4y2Ba
HMP1-II数据集还包括许多技术复制(252),这是有助于区分两个fast-varying组件(生物和技术噪声)。我们鼓励的一个非平凡的许多技术在未来的纵向研究,复制不只是为了验证还允许多样性的定量描述,并没有被纳入实验的其余部分由于有限的抽样率。因为技术噪声也与其他方差分量估计,估计的相对大小技术噪声也报道(gydF4y2Ba补充表5gydF4y2Ba)。由于技术噪声方差的比例通常是低的物种丰度(19.3%的中位数为5.4%,90)比途径(44%)的中位数为16%,90,与观测一致,真正的生物通路丰度低于物种丰度之间变化gydF4y2Ba1gydF4y2Ba。噪音水平途径主要是受体内网站,通路前鼻孔有最大的噪音(平均为40%)。gydF4y2Ba
我们评估的准确性参数拟合过程这些噪声条件下模拟样本混合物的三个组件和执行参数适合每个目标身体网站(gydF4y2Ba扩展数据图4 cgydF4y2Ba)。噪音水平,纯组件总是推断高信心,inter-individual差异是最可识别的。混合物inter-individual动力学与生物噪声也自信地恢复,而混合物inter-individual和生物噪声更变量,和混合物inter-individual和时变动力学是偏向时变动力学的影响更大。因此,当存在时变组件、参数估计应该考虑偏离inter-individual角落的三元图。所有三个组件最大的不确定性的混合物。在机构网站,推断前鼻孔和穹窿抽样分布是最不可靠的,因为在这些网站数量相对有限的样本(gydF4y2Ba扩展数据图4gydF4y2Ba),反映了大量的高度不确定特性在这些网站(gydF4y2Ba图3gydF4y2Ba)。在20%技术噪声(第90百分位的噪声分布的物种),参数估计降低明显,倾向于之前的均值(所有组件的混合物)。这因此导致低物种和通路倾向于对三元的中心定位图(gydF4y2Ba图3gydF4y2Ba)。gydF4y2Ba
我们注意到特定功能(微生物或途径丰富),每个非技术组件代表所有进程的总和与颞签名影响特性,这些不一定反映内在的属性特征。外在的例子过程可能产生生物噪声包括,日常饮食差异,样本收集的时间相对于吃饭,刷牙和其他个人卫生,空间变异的微生物在主题(例如,梯度在凳子上),和周末/工作日的差异。外在inter-individual来源差异可能源于文化/种族(民族与几种微生物的丰度密切相关gydF4y2Ba1gydF4y2Ba)、习惯的差异(例如,习惯性的和罕见的牙齿刷子和拥有),和长期的饮食差异,等等。最后,时变过程可能包括属性,如体重或缓慢变化在饮食偏好。gydF4y2Ba
高斯过程参数优化细节gydF4y2Ba
所有的参数和模型比较适合进行通过获得GPstuff在MATLAB工具箱版本4.6。拟合之前,相对丰度第一次平方根反正弦转换、过滤为离群值异常值使用Grubbs测试(0.05意义阈值),零均值和单位方差和标准化。与形状之前gamma-distributed 3.1和10个月是对lengthscale参数时变的组件。这些参数为gydF4y2BalgydF4y2Ba选择基于样本之间的间隔,并保证模型时可识别的生物噪声和/或组件包括通过确保inter-individual区别吗gydF4y2BalgydF4y2Ba既不能太短,也不能太长。所有模型的所有参数同时配合。所有模型都适合使用高斯可能性。这对高度非高斯分布函数执行差,经常发生在微生物数据zero-inflated丰度分布的形式。出于这个原因,进行了动态的分析高度普遍特性(物种≥75%患病率在网站,和核心通路)。一个例外是:物种的意思是丰富当出席≥2%,非零至少50个样本还包括,包括重要的物种,如gydF4y2Ba普氏菌coprigydF4y2Ba患病率较低,但当出现异常丰富。其他模型专门为零通胀会计(包括技术和实际)需要研究罕见的微生物的动态。gydF4y2Ba
出示的证据gydF4y2Ba补充表5gydF4y2Ba从5获得链/模型计算,150个样本20个样本老化后,都开始从先验分布的随机点。参数估计在gydF4y2Ba图3gydF4y2Ba和gydF4y2Ba补充表5gydF4y2Ba是符合额外的约束呢gydF4y2BaUgydF4y2Ba+gydF4y2BaTgydF4y2Ba+gydF4y2BaBgydF4y2Ba+gydF4y2BaNgydF4y2Ba= 1,消除额外自由度的模型。狄利克雷(1,1,1,1)之前是强加给gydF4y2BaUgydF4y2Ba,gydF4y2BaTgydF4y2Ba,gydF4y2BaBgydF4y2Ba,gydF4y2BaNgydF4y2Ba]。为每个特性测试,获得一个更全面的模型比模型选择,执行10链组成的200个样本(30老化和稀释其他样本),从一个随机的先验分布的点。同时在所有情况下,所有参数都符合。收敛性的评估gydF4y2Ba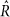 统计gydF4y2Ba48gydF4y2Ba。所有196种和950通道测试,97%的gydF4y2Ba
统计gydF4y2Ba48gydF4y2Ba。所有196种和950通道测试,97%的gydF4y2Ba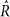 统计是< 1.1的所有参数(平均1.01,最大值1.17),表明良好的收敛性。gydF4y2Ba
统计是< 1.1的所有参数(平均1.01,最大值1.17),表明良好的收敛性。gydF4y2Ba
协会协变量微生物功能和表型之间的测试gydF4y2Ba
微生物之间的关联并使用MaAsLin通路丰度和元数据确定gydF4y2Ba1gydF4y2Ba,gydF4y2Ba49gydF4y2Ba。MaAsLin测试稀疏多变量广义线性模型独立对每个特性。相对丰度第一次平方根反正弦转换为方差稳定,使用和Grubbs测试(0.05显著性水平)删除离群值。运用单变量预选使用提高识别潜在的关联特性,和协变量显著相关剩余功能被确定与一个没有零通胀的多元线性模型。除非另有说明,最后罗斯福< 0.1 (Benjamini-Hochberg控制功能测试)被用作意义阈值。gydF4y2Ba
相同的模型应用于所有功能(微生物和通路)在此分析和协变量包括以下:广泛的饮食特征,主体是否母乳喂养,温度、入口pH值,后穹窿pH值,性别,年龄,种族,研究天处理,测序中心临床中心的质量基础,人类阅读的比例,收缩压、舒张压、脉搏、是否生了主题,HMP1 / HMP1-II和BMI。这些元数据可以发现的摘要gydF4y2Ba扩展数据如表1gydF4y2Ba。值得注意的是,一些最近发现了混杂因素如交通时间gydF4y2Ba8gydF4y2Ba期间收集的粪便样本没有抽样。gydF4y2Ba
基准测试和组装协议设计gydF4y2Ba
我们几个汇编包括IDBA-UD相比gydF4y2Ba38gydF4y2Ba,MetaVelvetgydF4y2Ba50gydF4y2Ba,SOAPDenovo2gydF4y2Ba51gydF4y2Ba瑞士巴塞尔,贝尔(罗氏)射线gydF4y2Ba52gydF4y2Ba、黑桃gydF4y2Ba53gydF4y2Ba,和天鹅绒gydF4y2Ba54gydF4y2Ba使用八个样本(SRS017820 SRS014126、SRS052668 SRS017820, SRS048870, SRS020220, SRS057205和SRS017820)在五个身体网站代表一系列宏基因组的复杂性。的基础上组装尺寸、平均长度,分段水平,和将军长度,我们选择IDBA-UD处理所有HMP1-II样品。gydF4y2Ba
数字正常化gydF4y2Ba
质量控制后,为每个样本序列读取运行通过“数字正常化”管道之前组装。这个过程是为了减少,尽可能从最主要来源分类单元的体积信息(在不牺牲组装剩下的能力)含量分类单元可以组装更均匀,而不是他们的汇编软件读取丢弃的不充分覆盖(主要类群相比)。gydF4y2Ba
中位数gydF4y2BakgydF4y2Ba- m覆盖率首次估计为所有使用高棉Python库读取gydF4y2Ba55gydF4y2Ba。这些数据被用于过滤输入读取正常化gydF4y2BakgydF4y2Ba- m覆盖在预选的范围内:为每个gydF4y2BakgydF4y2Ba- m的20个核苷酸长度在每一个阅读,观察的总数gydF4y2BakgydF4y2Ba使用- m作为报道的一个代理。读取的值gydF4y2BakgydF4y2Ba- m覆盖率已经超过20人丢弃。剩余的读取是单份的削减在第一个实例gydF4y2BakgydF4y2Ba- m(代表假定的误差序列)。读取post-trim长度小于gydF4y2BakgydF4y2Ba- m长度(20核苷酸)也被丢弃。幸存的读取再次削减,这一次的在第一个实例丰富(> 50×)gydF4y2BakgydF4y2Ba- m;再次读取post-trim长度小于20核苷酸的丢弃。对于剩余的阅读,我们可以(基于中值gydF4y2BakgydF4y2Ba- m覆盖在第一步)删除所有读取的值gydF4y2BakgydF4y2Ba- m覆盖> 5×。这是一个更激进的过滤推定地冗余序列,在消除初始读取highly-overrepresented(冗余)gydF4y2BakgydF4y2Ba即或严重不足(错误)gydF4y2BakgydF4y2Ba即。gydF4y2Ba
为后续组装后质量控制和规范化,我们增加了gydF4y2BakgydF4y2Ba32个核苷酸(其余读取灵敏度最大化)和建造所有剩余的重叠图读取。这张图被划分为一组读的可能性高的内部重叠,分离组件在预先计算的“stoptags”:gydF4y2BakgydF4y2Ba- m序列在初始分析扫描自动识别的红色不可靠assembly-traversal节点。从每个这样的分区读取被提取到单独的FASTA文件。每个分区测试更多的子组,从最开始一致(排名的顺序图可分性)。Re-partitioning进行如上,但更激进的参数:stoptags initially-computed重叠图中明确检测和去除Re-partitioning之前(包括生成新stoptags切除后剩余的图前的)。一致读组分为子分区到底曾经用这种方法:进一步迭代风险过度拟合,并不能保证收敛到一个有意义的结果。gydF4y2Ba
IDBA-UD组装和后处理gydF4y2Ba
数字正常化后,每个最后分区与IDBA-UD独立于其他组件的组装。的值gydF4y2BakgydF4y2Ba在(20、30、40岁,…,80), IDBA-UD will attempt to assemble its partition (via de Bruijn graph methods) usingkgydF4y2Ba事情的大小gydF4y2BakgydF4y2Ba,然后合并和扩展的结果通过生产总装的分区(100个核苷酸的要求最小重叠群长度)。对于每一个样本(或池),所有(独立)分区程序集被连接。作为最后一步,以减少任何冗余礼物在最后的连接装配,我们合并和扩展重叠群装配(所有分区),基于重叠40核苷酸或更多,产生最终的“合并”序列集合。gydF4y2Ba
质量评估gydF4y2Ba
评估装配质量,我们进行了一系列post-assembly质量控制检查,包括读取的速度一致的检查程序集以及识别嵌合体,这是一个潜在的问题由mis-assemblies引起的。gydF4y2Ba
检查部分读取所纳入大会,读取样本一致反对他们组装使用领结v1,导致计数与至少一个读取校准和对那些未能对齐。从人类宿主总读包括读。因为人类读取被使用BMTagger SRA,掩盖了所有Ns的人读起来会影响部分对齐。评估其效果,我们清点的数量蒙面读取获得人类读取一个计数。这些由身体网站进行了总结gydF4y2Ba扩展数据图7 cgydF4y2Ba。gydF4y2Ba
装配协议验证gydF4y2Ba
检查的空想的叠连群mis-assemblies,我们进行了一个装配评估2模拟数据集生成高分子聚合物中,创建一个社区与所有21生物丰度相等(“甚至”),和一个与交错丰度。我们组装这些模拟社区使用相同的协议和对齐重叠群装配对所有21输入基因组两组。我们发现,分别为94.21%和96.84%,所有组装叠连群对齐独特甚至一个参考基因组,staggered-coverage模拟社区(此处的“对齐”序列的身份与≥95%≥95%的长度)。叠连群一致密切相关gydF4y2Ba葡萄球菌gydF4y2Ba和gydF4y2Ba链球菌gydF4y2Ba菌株表现出非排他性匹配(或交叉配血)略高于其他菌株叠连群对齐。甚至,平均的97.85%gydF4y2Ba葡萄球菌gydF4y2Ba- - -gydF4y2Ba链球菌gydF4y2Ba对齐叠连群独特对齐参考应变时,平均92.98%的交错,所有其他菌株相比平均99.89%(甚至)和98.98%(交叉),巧妙地反映了内在的遗传模糊这些分类学的狭窄的子组表现出很强的能力区分相关的菌株。gydF4y2Ba
恢复统计不相关与交错设置输入覆盖,这意味着我们的管道(至少4×报道)强劲的反对的相对丰度的差异在这些尺度三个数量级。系统,在这种情况下,似乎显示出更大的影响力的独特性组装(尽管仍然非常薄弱)比报道。分数的重叠群不调整任何21参考菌株(≥95%的长度≥95%的身份)分别为5.6%和3.0%分别甚至和交错设置;我们可以假设这些比例结合率的上界嵌合体和mis-assemblies由我们的管道,与其他嵌合体装配指标一致gydF4y2Ba56gydF4y2Ba。gydF4y2Ba
注释gydF4y2Ba
检测并在重叠群装配使用metagenemark - 3.25执行gydF4y2Ba57gydF4y2Ba。由此产生的ORF序列作为输入对(1)UniRef100搜索gydF4y2Ba58gydF4y2Ba使用RAPSearch2gydF4y2Ba59gydF4y2Ba;(2)包含了gydF4y2Ba60gydF4y2Ba和TIGRfamgydF4y2Ba61年gydF4y2Ba嗯模型使用hmmer - 3.0gydF4y2Ba62年gydF4y2Ba;(3)TMHMMgydF4y2Ba63年gydF4y2Ba对跨膜螺旋的识别;和(4)一个正则表达式搜索膜脂蛋白脂质附件网站为公认的信号肽的识别。后者三个搜索Ergatis工作流监控系统中实现gydF4y2Ba64年gydF4y2Ba。gydF4y2Ba
注释以Attributor被分配(gydF4y2Bahttps://github.com/jorvis/AttributorgydF4y2Ba)使用分层方案开发的IGS原核的注释管道gydF4y2Ba65年gydF4y2Ba。Attributor分配共同的名字,基因符号、酶委员会(EC)数字和基因本体论(去)条款,如适用,基于证据包括支安打HMM模型的层次结构,UniRef100序列,TMHMM预测斜跨,脂蛋白图案。作业独占,这意味着每个ORF, Attributor需要可用的最强有力的证据和分配所有属性可能基于证据。属性不是来自多个源的分配,以确保注释属性分配给一个ORF不冲突。Attributor注释是输出gff3和FASTA文件(gydF4y2Ba扩展数据表1 bgydF4y2Ba)。gydF4y2Ba
稀疏曲线gydF4y2Ba
稀疏曲线生成的提取预测多肽MetaGeneMark输出为每个样本,和评估一个独特的基因家族的计数纯净的样本大小gydF4y2BangydF4y2Ba如下所示,使用usearch v.8.1.1861 x64gydF4y2Ba66年gydF4y2Ba:(1)连接MetaGeneMark预测多肽的随机抽样gydF4y2BangydF4y2Ba样品没有技术复制,消除重复;(2)通过减少排序序列长度;(3)集群序列在90%身份(使用usearch cluster_fast);(4)检索的独特基因家族的计数结果。独特的集群的数量估计来自50个随机子集gydF4y2BangydF4y2Ba。这个过程被重复每个网站gydF4y2BangydF4y2Ba= 1,10,20,…直到独特的样本的数量可以在身体的网站。gydF4y2Ba
读取映射到参考基因组gydF4y2Ba
除了分类和功能分析如上,所有样本的个体原始读取直接向MetaRef对齐gydF4y2Ba42gydF4y2Ba参考基因组。在校准之前,所有读取为80%或更高比例的Ns废弃使用Biocode fastq:: filter_fastq_by_N_content实用程序(gydF4y2Bahttps://github.com/jorvis/biocode/blob/master/fastq/filter_fastq_by_N_content.pygydF4y2Ba)。Bowtie2gydF4y2Ba67年gydF4y2Ba(v2.2.4)被用来使读取使用默认的参考基因组,paired-end对齐选项,包括单读取。由此产生的山姆文件转换为BAM,排序,然后划分为两个单独的文件中每个样本——一个只有匹配的读取和其他未对齐的读取。整个管道封装在Biocode generate_read_to_metaref_seed_alignment。py脚本(管道gydF4y2Bahttps://github.com/jorvis/biocode/blob/master/sandbox/jorvis/generate_read_to_metaref_seed_alignment.pygydF4y2Ba)。gydF4y2Ba
映射读取重叠群装配gydF4y2Ba
quality-trimmed读取从每个样本被映射到组装叠连群从相同的示例使用领结(v0.12.9)和一个512 MB马克斯最佳优先搜索帧值,Phred33质量分数设置,21个碱基对种子长度,并限制每种子2不匹配。校准每读都是报道(除非有超过20对于一个给定的读)达到保证最好的地层和质量。在次优地层没有报道。gydF4y2Ba
代码的可用性gydF4y2Ba
代码注释可从管道和高斯过程分析gydF4y2Ba扩展数据表1 bgydF4y2Ba。gydF4y2Ba
数据可用性gydF4y2Ba
序列数据可从HMP通道(gydF4y2Bahttp://hmpdacc.orggydF4y2Ba)或在亚马逊(gydF4y2Bahttps://aws.amazon.com/datasets/human-microbiome-project/gydF4y2Ba);WMS读取和相应的元数据是可用的序列读取存档(SRA;gydF4y2Bahttps://www.ncbi.nlm.nih.gov/sragydF4y2Ba)和数据库的基因型和表型(dbGaP;gydF4y2Bahttps://www.ncbi.nlm.nih.gov/gapgydF4y2Ba)在两个研究:gydF4y2BaSRP002163gydF4y2Ba(BioProjectgydF4y2BaPRJNA48479gydF4y2Ba),gydF4y2BaSRP056641gydF4y2Ba(BioProjectgydF4y2BaPRJNA275349gydF4y2Ba)。公共和私人的元数据gydF4y2Ba扩展数据表1gydF4y2Ba可用的宏基因组丰度分类单元表HMP通道(gydF4y2Bahttps://www.hmpdacc.org/hmsmcp2/gydF4y2Ba),并通过dbGaP加入phs000228.v3数量。分别p1。所有其他数据可从相应的作者在合理的请求。gydF4y2Ba
加入代码gydF4y2Ba
主要登记入册gydF4y2Ba
BioProjectgydF4y2Ba
顺序读取存档gydF4y2Ba
改变历史gydF4y2Ba
2017年10月12日gydF4y2Ba
大自然550年,61 - 66 (2017);doi: 10.1038 / nature23889这篇文章应该包含一个相关的知识共享声明作者信息部分。这是文章的在线版本中更正。gydF4y2Ba
引用gydF4y2Ba
人类微生物组项目财团。结构、功能和人类健康的微生物多样性。gydF4y2Ba自然gydF4y2Ba486年gydF4y2Ba207 - 214 (2012)gydF4y2Ba
罗伊德•普莱斯,J。,一个bu-Ali, G. & Huttenhower, C. The healthy human microbiome.基因组医学。gydF4y2Ba8gydF4y2Ba51 (2016)gydF4y2Ba
Gensollen, T。艾耶S。,Kasper, D. L. & Blumberg, R. S. How colonization by microbiota in early life shapes the immune system.科学gydF4y2Ba352年gydF4y2Ba539 - 544 (2016)gydF4y2Ba
本田、k .李曼荣& d . r .适应性免疫内稳态和疾病的微生物群。gydF4y2Ba自然gydF4y2Ba535年gydF4y2Ba75 - 84 (2016)gydF4y2Ba
秦,j . et al。人类肠道微生物基因目录建立了宏基因组测序。gydF4y2Ba自然gydF4y2Ba464年gydF4y2Ba59 - 65 (2010)gydF4y2Ba
李,j . et al .集成目录参考基因在人类肠道微生物组。gydF4y2Ba生物科技Nat。》。gydF4y2Ba32gydF4y2Ba834 - 841 (2014)gydF4y2Ba
博蒙特,m . et al。人类粪便微生物遗传组成部分与内脏脂肪有关。gydF4y2Ba基因组医学杂志。gydF4y2Ba17gydF4y2Ba189 (2016)gydF4y2Ba
Falony, g . et al .肠道微生物群体分析变异。gydF4y2Ba科学gydF4y2Ba352年gydF4y2Ba560 - 564 (2016)gydF4y2Ba
Zhernakova, a . et al。基于宏基因组分析显示标记为肠道微生物组成和多样性。gydF4y2Ba科学gydF4y2Ba352年gydF4y2Ba565 - 569 (2016)gydF4y2Ba
是的,J。,You, H. J., Yu, J., Sung, J. & Ko, G.普氏菌gydF4y2Ba作为阴道微生物群的中心的影响下宿主基因及其与肥胖协会。gydF4y2Ba细胞宿主细菌gydF4y2Ba21gydF4y2Ba97 - 105 (2017)gydF4y2Ba
冈萨雷斯,a . et al .偏头痛是与更高水平的硝酸盐,亚硝酸盐和硝酸oxide-reducing口服肠道微生物在美国项目群。gydF4y2BamSystemsgydF4y2Bahttps://doi.org/10.1128/mSystems.00105-16gydF4y2Ba(2016)gydF4y2Ba
哦,j . et al。生物地理学和个性在人类皮肤metagenome形状函数。gydF4y2Ba自然gydF4y2Ba514年gydF4y2Ba59 - 64 (2014)gydF4y2Ba
人类微生物组项目财团。人类微生物组研究的框架。gydF4y2Ba自然gydF4y2Ba486年gydF4y2Ba215 - 221 (2012)gydF4y2Ba
Truong, d . T。邰蒂,。,P一个年代olli, E., Huttenhower, C. & Segata, N. Microbial strain-level population structure and genetic diversity from metagenomes.基因组Res。gydF4y2Ba27gydF4y2Ba626 - 638 (2017)gydF4y2Ba
Schloissnig, s . et al .基因组变异人类肠道微生物组的景观。gydF4y2Ba自然gydF4y2Ba493年gydF4y2Ba45 - 50 (2013)gydF4y2Ba
罗,c . et al .约束识别微生物菌株在宏基因组数据集。gydF4y2Ba生物科技Nat。》。gydF4y2Ba33gydF4y2Ba1045 - 1052 (2015)gydF4y2Ba
木村,m .一个简单的方法来估计进化的基础替换率通过核苷酸序列的比较研究。gydF4y2Baj·摩尔。另一个星球。gydF4y2Ba16gydF4y2Ba111 - 120 (1980)gydF4y2Ba
Franzosa, e·a . et al。确定个人使用的微生物宏基因组编码。gydF4y2BaProc。《科学。美国gydF4y2Ba112年gydF4y2BaE2930-E2938 (2015)gydF4y2Ba
Leinonen, R。,年代ug一个wara, H. & Shumway, M. The sequence read archive.核酸Res。gydF4y2Ba39gydF4y2BaD19-D21 (2011)gydF4y2Ba
Truong, d . t . et al . MetaPhlAn2增强metagenomic分类分析。gydF4y2BaNat方法。gydF4y2Ba12gydF4y2Ba902 - 903 (2015)gydF4y2Ba
霍夫曼,c . et al。人类肠道微生物组的古菌和真菌:相关性与饮食和细菌的居民。gydF4y2Ba《公共科学图书馆•综合》gydF4y2Ba8gydF4y2Bae66019 (2013)gydF4y2Ba
Schwiertz, a . et al .微生物群和SCFA精益和超重的健康受试者。gydF4y2Ba肥胖gydF4y2Ba18gydF4y2Ba190 - 195 (2010)gydF4y2Ba
骄傲,d . t . et al .噬菌体常住人口强劲的证据显示人类唾液virome通过分析。gydF4y2BaISME J。gydF4y2Ba6gydF4y2Ba915 - 926 (2012)gydF4y2Ba
Abubucker, s . et al .代谢重建人类微生物宏基因组数据及其应用。gydF4y2Ba公共科学图书馆第一版。医学杂志。gydF4y2Ba8gydF4y2Bae1002358 (2012)gydF4y2Ba
卡斯皮,r . et al . MetaCyc数据库和酶的代谢通路和BioCyc通路/基因组数据库的集合。gydF4y2Ba核酸Res。gydF4y2Ba42gydF4y2BaD459-D471 (2014)gydF4y2Ba
Leonardi, R。,Zhang, Y. M., Rock, C. O. & Jackowski, S. Coenzyme A: back in action.掠夺。脂质物。gydF4y2Ba44gydF4y2Ba125 - 153 (2005)gydF4y2Ba
Khakh b . s . & Burnstock g . ATP的双重生活。gydF4y2Ba科学。点。gydF4y2Ba301年gydF4y2Ba84 - 92 (2009)gydF4y2Ba
Morkbak, a . L。,Poulsen, S. S. & Nexo, E. Haptocorrin in humans.中国。化学。实验室。地中海。gydF4y2Ba45gydF4y2Ba1751 - 1759 (2007)gydF4y2Ba
罗伊,C . C。,Kien, C. L., Bouthillier, L. & Levy, E. Short-chain fatty acids: ready for prime time?减轻。中国。Pract。gydF4y2Ba21gydF4y2Ba351 - 366 (2006)gydF4y2Ba
施赖伯,f . et al。反硝化作用在人类牙菌斑。gydF4y2BaBMC医学杂志。gydF4y2Ba8gydF4y2Ba24 (2010)gydF4y2Ba
弗林特,h·J。,年代cott, K. P., Duncan, S. H., Louis, P. & Forano, E. Microbial degradation of complex carbohydrates in the gut.肠道微生物gydF4y2Ba3gydF4y2Ba289 - 306 (2012)gydF4y2Ba
信仰,j。j。人类肠道微生物群的长期稳定。gydF4y2Ba科学gydF4y2Ba341年gydF4y2Ba1237439 (2013)gydF4y2Ba
弗洛雷斯,g . e . et al .颞可变性是人类微生物组的个性化特征。gydF4y2Ba基因组医学杂志。gydF4y2Ba15gydF4y2Ba531 (2014)gydF4y2Ba
叮,t &城堡,p . d .动力学和人体微生物群落类型的关联。gydF4y2Ba自然gydF4y2Ba509年gydF4y2Ba357 - 360 (2014)gydF4y2Ba
恩伯,p . j . et al .人类微生物组计划。gydF4y2Ba自然gydF4y2Ba449年gydF4y2Ba804 - 810 (2007)gydF4y2Ba
Shafquat,。乔伊斯,R。,年代我mmons, S. L. & Huttenhower, C. Functional and phylogenetic assembly of microbial communities in the human microbiome.Microbiol趋势。gydF4y2Ba22gydF4y2Ba261 - 266 (2014)gydF4y2Ba
Gajer, p . et al .颞动力学的人类阴道微生物群。gydF4y2Ba科学。Transl。地中海。gydF4y2Ba4gydF4y2Ba132 ra52 (2012)gydF4y2Ba
彭,Y。,Leung, H. C., Yiu, S. M. & Chin, F. Y. IDBA-UD: a新创gydF4y2Ba汇编单细胞和宏基因组测序数据的深度与高度不均匀。gydF4y2Ba生物信息学gydF4y2Ba28gydF4y2Ba1420 - 1428 (2012)gydF4y2Ba
芬恩,r . d . et al .蛋白质包含了家庭数据库:向一个更可持续的未来。gydF4y2Ba核酸Res。gydF4y2Ba44gydF4y2BaD279-D285 (2016)gydF4y2Ba
Aagaard, k等。人类微生物组计划策略综合抽样人类微生物组和为什么它很重要。gydF4y2Ba美国实验生物学学会联合会J。gydF4y2Ba27gydF4y2Ba1012 - 1022 (2013)gydF4y2Ba
Caporaso, j·g . et al . QIIME允许社区高通量测序数据的分析。gydF4y2BaNat方法。gydF4y2Ba7gydF4y2Ba335 - 336 (2010)gydF4y2Ba
黄,k . et al . MetaRef: pan-genomic数据库比较微生物基因组学和社区。gydF4y2Ba核酸Res。gydF4y2Ba42gydF4y2BaD617-D624 (2014)gydF4y2Ba
Suzek b E。王,Y。,Huang, H., McGarvey, P. B. & Wu, C. H. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches.生物信息学gydF4y2Ba31日gydF4y2Ba926 - 932 (2015)gydF4y2Ba
卡斯皮,r . et al . MetaCyc数据库和酶的代谢通路和BioCyc通路/基因组数据库的集合。gydF4y2Ba核酸Res。gydF4y2Ba44gydF4y2BaD471-D480 (2016)gydF4y2Ba
加尔佩林,m . Y。,Makarova, K. S., Wolf, Y. I. & Koonin, E. V. Expanded microbial genome coverage and improved protein family annotation in the COG database.核酸Res。gydF4y2Ba43gydF4y2BaD261-D269 (2015)gydF4y2Ba
爸爸,t . et al .建设gydF4y2Ba大肠杆菌gydF4y2Bak - 12在坐标系,单基因敲除突变体:庆应义塾集合。gydF4y2Ba摩尔。系统。医学杂志。gydF4y2Ba2gydF4y2Ba2006.0008 (2006)gydF4y2Ba
UniProt财团。UniProt:通用的蛋白质知识库。gydF4y2Ba核酸Res。gydF4y2Ba45gydF4y2BaD158-D169 (2017)gydF4y2Ba
> a &鲁宾,d . b .推理从迭代模拟使用多个序列。gydF4y2Ba统计科学。gydF4y2Ba7gydF4y2Ba457 - 472 (1992)gydF4y2Ba
摩根,x c . et al .肠道微生物的功能障碍炎性肠道疾病和治疗。gydF4y2Ba基因组医学杂志。gydF4y2Ba13gydF4y2BaR79 (2012)gydF4y2Ba
只,T。,Hachiya, T., Tanaka, H. & Sakakibara, Y. MetaVelvet: an extension of Velvet assembler to新创gydF4y2Ba从短序列读取metagenome组装。gydF4y2Ba核酸Res。gydF4y2Ba40gydF4y2Bae155 (2012)gydF4y2Ba
罗,r . et al . SOAPdenovo2:一个经验改进节约内存短内容gydF4y2Ba新创gydF4y2Ba汇编程序。gydF4y2BaGigaSciencegydF4y2Ba1gydF4y2Ba,18 (2012);勘误表4 1 (2015)gydF4y2Ba
布瓦维尔说,S。,Raymond, F., Godzaridis, E., Laviolette, F. & Corbeil, J. Ray Meta: scalable新创gydF4y2Bametagenome汇编和分析。gydF4y2Ba基因组医学杂志。gydF4y2Ba13gydF4y2BaR122 (2012)gydF4y2Ba
Bankevich, a . et al .黑桃:一个新的基因组组装算法及其应用单细胞测序。gydF4y2Baj .第一版。医学杂志。gydF4y2Ba19gydF4y2Ba455 - 477 (2012)gydF4y2Ba
Zerbino d r &伯尼大肠天鹅绒:算法gydF4y2Ba新创gydF4y2Ba短阅读使用de Bruijn图组装。gydF4y2Ba基因组Res。gydF4y2Ba18gydF4y2Ba821 - 829 (2008)gydF4y2Ba
佩尔,j . et al .缩放metagenome序列组装与概率de Bruijn图表。gydF4y2BaProc。《科学。美国gydF4y2Ba109年gydF4y2Ba13272 - 13277 (2012)gydF4y2Ba
曼德,d . r . et al .评估宏基因组组装使用模拟下一代测序数据。gydF4y2Ba《公共科学图书馆•综合》gydF4y2Ba7gydF4y2Bae31386 (2012)gydF4y2Ba
朱,W。,Lomsadze, A. & Borodovsky, M.从头开始gydF4y2Ba基因识别宏基因组序列。gydF4y2Ba核酸Res。gydF4y2Ba38gydF4y2Bae132 (2010)gydF4y2Ba
Suzek b E。黄,H。,McGarvey, P., Mazumder, R. & Wu, C. H. UniRef: comprehensive and non-redundant UniProt reference clusters.生物信息学gydF4y2Ba23gydF4y2Ba1282 - 1288 (2007)gydF4y2Ba
赵,Y。,T一个ng,H. & Ye, Y. RAPSearch2: a fast and memory-efficient protein similarity search tool for next-generation sequencing data.生物信息学gydF4y2Ba28gydF4y2Ba125 - 126 (2012)gydF4y2Ba
芬恩,r . d . et al。包含:家庭的蛋白质数据库。gydF4y2Ba核酸Res。gydF4y2Ba42gydF4y2BaD222-D230 (2014)gydF4y2Ba
安顿下来,d . h . et al . 2013年TIGRFAMs和基因组特性。gydF4y2Ba核酸Res。gydF4y2Ba41gydF4y2BaD387-D395 (2013)gydF4y2Ba
艾迪,s . r .加速概要嗯搜索。gydF4y2Ba公共科学图书馆第一版。医学杂志。gydF4y2Ba7gydF4y2Bae1002195 (2011)gydF4y2Ba
Sonnhammer, e . L。,von Heijne, G. & Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences.Proc, Int。相依智能。系统。摩尔。杂志。gydF4y2Ba6gydF4y2Ba175 - 182 (1998)gydF4y2Ba
Orvis j . et al . Ergatis:一个web界面和可伸缩的生物信息学软件系统工作流程。gydF4y2Ba生物信息学gydF4y2Ba26gydF4y2Ba1488 - 1492 (2010)gydF4y2Ba
盖伦,k . et al . IGS标准操作程序进行自动原核的注释。gydF4y2Ba的立场。基因组科学。gydF4y2Ba4gydF4y2Ba244 - 251 (2011)gydF4y2Ba
埃德加,r . c .搜索和集群数量级的速度比爆炸。gydF4y2Ba生物信息学gydF4y2Ba26gydF4y2Ba2460 - 2461 (2010)gydF4y2Ba
Langmead, b &扎尔茨贝格,s . l .快gapped-read符合领结2。gydF4y2BaNat方法。gydF4y2Ba9gydF4y2Ba357 - 359 (2012)gydF4y2Ba
Roager, h . m . et al .结肠转运时间与细菌新陈代谢和肠道粘膜营业额。gydF4y2BaMicrobiol Nat。gydF4y2Ba1gydF4y2Ba16093 (2016)gydF4y2Ba
确认gydF4y2Ba
我们承认j . a . Aluvathingal g . Shankar和k . Shefchek贡献在本项目运行分析,诉Felix让数据可以通过数据分析和协调中心(通道)门户网站,原HMP外部科学顾问委员会(r . Blumberg j·戴维斯,r·霍尔特·Ossorio f . Ouellette) g . Schoolnik和a . Williamson),在整个国际合作者人类微生物组的财团,和个人的参与从圣路易斯,密苏里州和得克萨斯州休斯敦的地区使人类微生物组计划成为可能。这项工作是由美国国立卫生研究院(NIH)赠款U54DK102557 h,U01HG006537 U01HG004866 O.W.,U54HG004969 to B. W. Birren, U54HG004973 to R. A. Gibbs, S. K. Highlander and J. F. Petrosino, U54AI084844 to K. E. Nelson, U54HG004968 to G. M. Weinstock, U54HG003079 to R. K. Wilson, U01HG006537, R01HG004872, and U01HG004866 to R.K., and Crohn’s & Colitis Foundation of America (CCFA) award no. 3162 to J. Braun with subaward to C.H. and R. Xavier.
作者信息gydF4y2Ba
作者和联系gydF4y2Ba
贡献gydF4y2Ba
广义相对论和点收集和组织数据。广义相对论生成和分析分类和(E.A.F.)和主机功能配置元数据关联。J.L.-P。设计和执行高斯过程模型。点,M.G.G., H.H.C., J.C., J.O., C.M. and A.B. performed assembly and annotation. J.L.-P., G.R., E.A.F., B.H., A.M., A.B., M.G.G., O.W. and C.H. drafted the manuscript. C.H., O.W. and R.K. designed the study. All authors discussed the results and commented on the paper.
相应的作者gydF4y2Ba
道德声明gydF4y2Ba
相互竞争的利益gydF4y2Ba
作者声明没有竞争的经济利益。gydF4y2Ba
额外的信息gydF4y2Ba
审核人信息gydF4y2Ba自然gydF4y2Ba由于h·b·尼尔森,p .恩伯,另一个匿名的评论家(s)为他们的贡献的同行评审工作。gydF4y2Ba
出版商的注意:施普林格自然保持中立在发表关于司法主权地图和所属机构。gydF4y2Ba
扩展数据数据和表gydF4y2Ba
图1扩展数据扩展全身宏基因组HMP1-II分类资料。gydF4y2Ba
一个gydF4y2Ba,结合HMP1-II数据集包括2355基因组(724年以前和1631年出版新,包括252年技术复制)。这些跨项目的六个目标身体网站(前鼻孔,颊粘膜,supragingival斑块,舌背,凳子上,和穹窿)除了至少20个样品各3额外的网站,18岁的总采样地点:retroauricular折痕,腭扁桃体,龈下的斑块。基因组现在用于至少一个身体网站共有265人。gydF4y2BabgydF4y2Ba,PCoA使用Bray-Curtis距离在所有微生物在物种水平。gydF4y2BacgydF4y2Ba,最普遍的相对丰度和丰富的微生物(细菌、病毒、真核和热点)在所有网站,由MetaPhlAn2异形gydF4y2Ba20.gydF4y2Ba。普遍的真核微生物在属级显示。gydF4y2BadgydF4y2Ba分类,测序中心之间,不同批次,或临床中心比个体身体内网站。天秀Bray-Curtis了解的主要坐标丰度在每个站点。网站或生态结构gydF4y2Ba1gydF4y2Ba,没有散度与技术相关的变量在前两个配合的轴。gydF4y2Ba
扩展数据图2地理、时间和生物地理的应变变化。gydF4y2Ba
一个gydF4y2Ba之间的平均距离(木村两个参数)菌株从对象内部或之间的三位邮政编码(地理信息的最好的程度)。数据和样本大小gydF4y2Ba补充表2gydF4y2Ba。gydF4y2BabgydF4y2Ba,平均应变之间的差异不同访问相同的主题和正文的网站相比,之间的平均距离相同的访问同样的主题和正文的网站为每个物种(技术复制)。gydF4y2BacgydF4y2Ba- - - - - -gydF4y2BaugydF4y2Ba木村,PCoA情节基于两个参数的距离gydF4y2Ba17gydF4y2Ba所示为gydF4y2Ba大肠杆菌gydF4y2Ba(gydF4y2BacgydF4y2Ba),gydF4y2Ba放线菌johnsoniigydF4y2Ba(gydF4y2BadgydF4y2Ba和所有物种所示gydF4y2Ba图1 bgydF4y2Ba;gydF4y2BaegydF4y2Ba- - - - - -gydF4y2BaugydF4y2Ba),按照降序排列的niche-association得分(方法)。距离矩阵用于生成这些PCoAs公开(gydF4y2Ba扩展数据表1 bgydF4y2Ba)。gydF4y2Ba
扩展数据图3网站人体微生物组的核心和区分功能。gydF4y2Ba
这个数字扩展gydF4y2Ba图3gydF4y2Ba,更多的细节和例子。gydF4y2Ba一个gydF4y2Ba28岁的代谢途径是核心(普遍> 75%)所有主要机构网站。我们称这些为“supercore”途径。gydF4y2BabgydF4y2Ba通路核心身体更多的网站往往有更广泛的分类范围,与supercore通路中最广泛分布(图基箱线图)。gydF4y2BacgydF4y2Ba,19个通道(包括两个supercore途径,出演gydF4y2Ba一个gydF4y2Ba)在多个身体核心区域和特别丰富类群存在于人类微生物组(注释< non-human-associated属的10%)。Human-microbiome-enriched通路包括特定的更广泛的定义或分布式MetaCyc-defined变异过程,例如,肽聚糖生物合成(pwy - 6471)。gydF4y2BadgydF4y2Ba,Site-enriched路径在一个身体更加丰富网站比网站从身体其他地区。黑点表示该网站每个site-enriched途径达到顶峰。热图值反映了相对丰富的第一四分位数在身体特定站点(协调的百分比截止核心途径)。gydF4y2BaegydF4y2Ba- - - - - -gydF4y2BaggydF4y2Ba,额外的三个途径类枚举的例子gydF4y2Ba一个gydF4y2Ba,gydF4y2BacgydF4y2Ba,gydF4y2BadgydF4y2Ba,分别。在每个例子中,总(社区)富足是对数,前7个属的贡献比例缩放在社区内。“其他”包含之外的途径来自属前七,和“非机密”包括社区的途径来自不明身份的成员。gydF4y2Ba
图4扩展数据采样间隔分布,参数适合模拟样品和微生物物种丰度的动态和对应的高斯过程符合。gydF4y2Ba
一个gydF4y2Ba样本之间的时间差异,分布在每一个有针对性的身体。技术复制显示为ΔgydF4y2BatgydF4y2Ba= 0。gydF4y2BabgydF4y2Ba、参数适合模拟样品gydF4y2BaUgydF4y2Ba= 0,gydF4y2BaBgydF4y2Ba= 0,gydF4y2BaTgydF4y2Ba= 0.95,gydF4y2BaNgydF4y2Ba= 0.05,不同gydF4y2BalgydF4y2Ba(见方法)。模拟样本的实际样本分布,从每个站点数,显示限制在某些网站改变抽样的忠诚。gydF4y2BacgydF4y2Ba、参数适合5模拟样品的三个纯组件(颜色红色、绿色和蓝色),以及对他们的所有甚至混合物(例如,黄色点甚至混合物gydF4y2BaUgydF4y2Ba和gydF4y2BaTgydF4y2Ba),甚至三(黑)的混合物,不同层次的技术噪声(gydF4y2BaNgydF4y2Ba)和固定gydF4y2BalgydF4y2Ba= 0.5。不确定推理更去饱和。gydF4y2BadgydF4y2Ba- - - - - -gydF4y2BafgydF4y2Ba,三个例子分类资料符合高斯过程模型显示在情节设计允许数据和之间的直接比较符合高斯过程,并允许不同的动态可视化尽管限制每人只有三个时间点。每个例子是选择的范例模型中的三个非技术组件之一。Insets表示信心十分位数获得的样本。丰富的gydF4y2Ba梭菌属periodonticumgydF4y2Ba在舌背显示强时变行为(gydF4y2BadgydF4y2Ba),gydF4y2Ba拟杆菌stercorisgydF4y2Ba在凳子上显示主要inter-individual差异(gydF4y2BaegydF4y2Ba),gydF4y2BaGemella haemolysansgydF4y2Ba在颊黏膜生物噪声是由(gydF4y2BafgydF4y2Ba)。情节显示绝对差的平方根反正弦转换微生物丰度(|ΔgydF4y2BaxgydF4y2Ba|)之间对同一个人的样本与样本之间的时差(点)。Gaussian-smoothed估计标准偏差的点也显示(蓝线,带宽三个月),以及期望的差异从符合高斯过程(红线)。标准差与Δ技术复制(点之间的差异gydF4y2BatgydF4y2Ba= 0个月)也显示为线存根在原点,直接可视化技术水平的噪音。生物噪声是可见的在这里技术之间的区别噪声和其余点的方差外推到原点。逐渐增加的时变组件是可见的差异的方差随时间(即逐渐增加红色和蓝色线)。最后,inter-individual差异可见通过比较的方差的极限数据与对象之间的差异的方差(绿线)。gydF4y2Ba
扩展数据图5高斯过程的时序方差分解宏基因组物种丰度。gydF4y2Ba
后的均值方差分解显示(方法)为每一个物种(gydF4y2Ba补充表5gydF4y2Ba),彩色的门。估计的不确定性评估通过均方距离的平方根的三元阴谋获得样本后的意思,并将与更大点指示更特定的估计。gydF4y2Ba
扩展数据图6组装注释为单身和co-assemblies特异性。gydF4y2Ba
一个gydF4y2Ba箱线图,图基的蛋白质的比例在每个功能特异性类别。gydF4y2BabgydF4y2Ba,例如维恩图一套single-sample supragingival斑块总成及其结合co-assembly (co-assembly在左下角),显示项共享基因(计算通过严格对齐)之间的所有程序集的组合;co-assembly本身包含所有检测到的基因的96.9%。gydF4y2BacgydF4y2Ba箱线图,图基数量的基因本体论(去)条款(生成使用苗条大约1700计算)之间共享单一和co-assemblies co-assembly独特,或独特的一个单一组件,生成的随机选择250名议会6身体网站。Co-assemblies捕捉方面,不是在单独的组件。gydF4y2Ba
图7扩展数据排序统计和装配质量评估。gydF4y2Ba
一个gydF4y2Ba,图基箱线图中每个样本的总生读的身体从SRA网站上检索。gydF4y2BabgydF4y2Ba,每具尸体百分比人类读着BMTagger网站。gydF4y2BacgydF4y2Ba的非人的百分比(细菌)读取对齐组件显示装配效率(方法;阅读和叠连群映射到程序集和参考基因组)。gydF4y2BadgydF4y2Ba,比较独特的领域包含的数量在每个样本中发现HUMAnN2总成,彩色的身体。域包含了HUMAnN2被认为是‘发现’如果UniRef50序列注释与域存在样本> 10每千碱基读取(约1×报道)。域包含的组件被检测出,如果他们被发现在一个叠连群以Attributor(方法)。gydF4y2BaegydF4y2Ba域包含了数量,发现至少有75%的样本(核心”域)每个方法,为每个目标网站。由未知函数包含域分层。gydF4y2Ba
扩展数据图8元基因组功能丰度明显与主机相关的表型。gydF4y2Ba
一个gydF4y2Ba,gydF4y2BabgydF4y2Ba,重要的协会重要的效应大小(罗斯福< 0.1和|gydF4y2BaβgydF4y2Ba| > 0.01)在多元线性模型(意义和系数gydF4y2Ba补充表5gydF4y2Ba分类单元之间的)丰度(gydF4y2Ba一个gydF4y2Ba)和通路丰度(gydF4y2BabgydF4y2Ba)。所有检测到的关联独立于所有其他元数据,包括主题是否母乳喂养,主体的广泛的饮食特征,温度、入口pH值,后穹窿pH值,性别,年龄,种族,研究天处理,测序中心临床中心,质量基础,人类阅读的比例,收缩压、舒张压、脉搏、是否生了主题,HMP1 / HMP1-II, BMI(集团大小gydF4y2Ba扩展数据表1gydF4y2Ba;见的方法)。与协会在这里不应被视为没有联系的证据。gydF4y2Ba
扩展数据图9 HMP1-II更新关联。gydF4y2Ba
一个gydF4y2BaHMP群组主题报道,他们是否母乳喂养的婴儿。值得注意的是,整体门壁厚菌门丰度较低甚至在成年期(受试者的当前年龄18-40)在个人历史母乳喂养。gydF4y2BabgydF4y2Ba,gydF4y2BacgydF4y2Ba,差异与其他婴儿母乳喂养持续演化支和身体网站,例如,口服gydF4y2Ba奈瑟氏菌属gydF4y2Ba(gydF4y2BabgydF4y2Ba),尽管age-linked关联不同类群之间(例如,整个口腔gydF4y2Ba奈瑟氏菌属gydF4y2Ba随着年龄增长而减少)(gydF4y2BacgydF4y2Ba)。gydF4y2BadgydF4y2Ba- - - - - -gydF4y2BafgydF4y2Ba,协会重要的例子在原始HMP1 metagenome集gydF4y2Ba1gydF4y2Ba保留的大HMP1-II数据集包括:gydF4y2BadgydF4y2Ba,gydF4y2Ba拟杆菌vulgatusgydF4y2Ba在凳子上明显更丰富的亚洲人相比其他种族的。gydF4y2BaegydF4y2Ba,gydF4y2Ba乳酸菌crispatusgydF4y2Ba后穹窿与阴道的pH值呈现负相关。gydF4y2BafgydF4y2Ba,gydF4y2Ba拟杆菌gydF4y2Ba更丰富的人的母乳喂养婴儿。箱线图胡须是由图基的方法。gydF4y2Ba
补充信息gydF4y2Ba
补充信息gydF4y2Ba
这个文件包含一个补充讨论和完整的传说为补充表1 - 11。(PDF 217 kb)gydF4y2Ba
补充表gydF4y2Ba
这个文件包含补充表1 - 11。(XLSX 703 kb)gydF4y2Ba
权利和权限gydF4y2Ba
这项工作是基于知识共享署名4.0国际4.0 (CC)许可证。本文中的图片或其他第三方材料都包含在本文的创作共用许可证,除非另有说明在信贷额度;如果材料不包括在Creative Commons许可下,用户需要获得许可执照持有人繁殖材料。查看本许可证的副本,访问gydF4y2Bahttp://creativecommons.org/licenses/by/4.0/gydF4y2Ba。gydF4y2Ba
关于这篇文章gydF4y2Ba

引用这篇文章gydF4y2Ba
罗伊德•普莱斯,J。补充一个。,Rahnavard, G.et al。gydF4y2Ba菌株、函数和动态扩展人类微生物组计划。gydF4y2Ba自然gydF4y2Ba550年gydF4y2Ba,61 - 66 (2017)。https://doi.org/10.1038/nature23889gydF4y2Ba
收到了gydF4y2Ba:gydF4y2Ba
接受gydF4y2Ba:gydF4y2Ba
发表gydF4y2Ba:gydF4y2Ba
发行日期gydF4y2Ba:gydF4y2Ba
DOIgydF4y2Ba:gydF4y2Bahttps://doi.org/10.1038/nature23889gydF4y2Ba
进一步的阅读gydF4y2Ba
Ontology-aware深度学习使超速和可翻译的源跟踪sub-million微生物群落样本数以百计的利基市场gydF4y2Ba
基因组医学gydF4y2Ba(2022)gydF4y2Ba
硫代谢基因的多样性和分布在人类肠道微生物及其与结直肠癌gydF4y2Ba
微生物组gydF4y2Ba(2022)gydF4y2Ba
孕产妇microbiota-derived代谢在胎鼠肠,大脑和胎盘gydF4y2Ba
BMC微生物学gydF4y2Ba(2022)gydF4y2Ba
宏基因组与SameStr应变检测:坚持核心的识别由粪便移植肠道微生物群的转移gydF4y2Ba
微生物组gydF4y2Ba(2022)gydF4y2Ba
微生物群的作用在重症监护病人的管理gydF4y2Ba
年报的重症监护gydF4y2Ba(2022)gydF4y2Ba
评论gydF4y2Ba
通过提交评论你同意遵守我们的gydF4y2Ba条款gydF4y2Ba和gydF4y2Ba社区指导原则gydF4y2Ba。如果你发现一些滥用或不符合我们的条件或准则请国旗是不合适的。gydF4y2Ba
