胰腺癌患者的唾液微生物的特性
- 发表
- 接受
- 收到了
- 学术编辑器
- 诺拉诺
- 版权
- ©2015年 托雷斯et al。
- 执照
- 这是一个开放的分布式根据文章知识共享归属许可,它允许无限制的使用、分配、繁殖和适应在任何媒介并为任何目的,只要它是正确的原因。归因的原始作者(年代),标题、出版来源(PeerJ)和DOI或文章的URL必须引用。
- 引用这篇文章
- 2015年。胰腺癌患者的唾液微生物的特性。PeerJ3:e1373 https://doi.org/10.7717/peerj.1373
文摘
胰腺癌的临床表现常不出现,直到癌症发生了转移,导致存活率很低。在这项研究中,我们调查是否唾液细菌谱可能提供有用的生物标记胰腺癌的早期检测。利用高通量测序的细菌小亚基核糖体RNA基因(16 s rRNA),我们与胰腺癌患者的唾液微生物群的特征,而他们健康的病人和其他疾病患者,包括胰腺疾病,non-pancreatic消化疾病/癌症和非消化道疾病癌症。总共146名患者在穆尔斯癌症中心UCSD的唾液和人口数据收集从每个病人。其中,我们分析了108例患者的唾液微生物:8被诊断出患有胰腺癌,78与其他疾病和22个被列为十几(健康)控制。细菌16 s rRNA直接从唾液提取DNA序列被放大并受高通量测序(高温超导)。一些细菌属不同丰度在胰腺癌患者。我们发现了一个显著的比例更高纤毛菌属来Porphyromonas在与胰腺癌患者的唾液健康患者的唾液或与其他疾病(克鲁斯卡尔-沃利斯检验;P< 0.001)。纤毛菌属使用实时qPCR与丰度被证实纤毛菌属特定的引物。类似于之前的研究,我们发现的相对丰度较低奈瑟氏菌属和Aggregatibacter在胰腺癌患者的唾液,尽管这些结果不显著P< 0.05级(知道测试;P= 0.07,P= 0.09)。然而,先前确定的相对丰度的其他细菌生物标记,例如,链球菌和Granulicatella adiacens没有显著不同的胰腺癌患者的唾液。总的来说,这项研究支持了假设唾液中细菌丰度的概要文件是有用的生物标记胰腺癌虽然还需要更大的患者的研究来验证他们的预测效用。
介绍
在美国,每年大约有40000人死于胰腺癌,使其成为第四个癌症相关死亡的主要原因。患者的早期诊断胰腺癌的5年生存率24%,1.8%在诊断高级阶段(李et al ., 2004)。胰腺癌的临床表现没有出现在癌症发生了转移(冬青et al ., 2004),强调早期发现生物标志物的必要性。胰腺癌的病因仍然是难以捉摸的,吸烟是最经常出现的风险因素(Vrieling et al ., 2010;中村et al ., 2011;福克斯,科迪兹&斯坦福,1996;郑et al ., 1993链接),尽管也取得了糖尿病(Haugvik et al ., 2015;刘et al ., 2015),肥胖(Bracci 2012)和慢性胰腺炎(无论et al ., 2002)。最近的研究也表明,牙周疾病的男性患胰腺癌的风险双重大调整后,吸烟,糖尿病,身体质量指数(米肖德et al ., 2007)。
人类口腔港口一个复杂的微生物群落(微生物)已知包含超过700种细菌,其中一半以上没有培养(Aas et al ., 2005)。研究人员已经确定了一个核心微生物群落在健康个体(Zaura Keijser &休斯,2009),从这个核心微生物与牙齿有关携带和牙周炎(Berezow & Darveau, 2011)。唾液中细菌群落的组成似乎反映健康状况在某些情况下(山中et al ., 2012),使唾液微生物的分析一个有前途的疾病诊断方法。的一项研究米塔尔et al。(2011)发现数量的增加变形链球菌和唾液中乳酸杆菌与口腔疾病患病率有关,而另一项研究表明,高唾项Capnocytophaga gingivalis,普氏菌melaninogenica和链球菌可能暗示口腔癌(马杰et al ., 2005)。
最近的一项研究法雷尔et al。(2012)建议具体唾液细菌的丰度可以作为生物标志物早期胰腺癌。使用人类的口腔微生物识别芯片(HOMIM),研究人员观察到的水平下降奈瑟氏菌属elongata和链球菌胰腺癌患者与健康人相比,而水平的Granulicatella adiacens在胰腺癌患者明显高于(法雷尔et al ., 2012)。HOMIM的能力来检测300种最常见的口腔细菌使得它一种适当的方法来评估社区档案在门级以及许多常见类群在属水平。然而,HOMIM微阵列方法无法检测到大约一半的细菌通常存在于唾液(安et al ., 2011)。
在这项研究中,我们应用高通量测序(高温超导)的细菌small-subunit核糖体RNA基因(16 s rRNA)来确定患者的唾液概要文件和胰腺癌。使用高温超导16 s rRNA细菌基因序列从整个唾液微生物群落允许更全面的健康和疾病的微生物(Kuczynski到来&沃尔特斯,2011)。在这项研究中,我们收集了146份唾液样本从病人UCSD穆尔斯癌症中心。高温超导被用来描述与胰腺癌患者的唾液微生物,并将它们与其他疾病患者比较(包括胰腺疾病、non-pancreatic消化疾病/癌症和非消化道疾病/癌症)以及十几(健康)控制。这允许我们测试的假设胰腺癌患者可能有不同的微生物群落配置文件比十几控制和其他形式的消化和非消化道疾病。我们的研究结果表明,胰腺癌患者有一个特定的相对丰度明显高于细菌属。
材料和方法
样本收集和患者信息
本研究通过加州大学圣地亚哥分校(UCSD)和圣地亚哥州立大学(圣地亚哥大学)联合机构审查委员会(IRB # 120101)批准。患者招募UCSD的研究被临床评估穆尔斯癌症中心或内镜手术由UCSD肠胃科桑顿医院Pre-Procedure诊所2012年5月到2013年8月。所有患者被要求禁食12小时前癌症评价和内窥镜检查程序。为了避免偏见在招生期间,研究协调员负责招聘参与者不知道病人诊断时样本集合。自愿参与者提供IRB-approved同意书,HIPAA形式,以及一个可选的、自愿的书面调查,他们可以分享关于抗生素的相关信息,牙科和吸烟史。所有参与者给予知情同意和他们的身份被扣留的研究团队。每个主题是自由随时退出研究。参与者被要求给一个唾液样本到50毫升锥形管。如果唾液的数量超过了55 uL, 10 uL转移到含有脑心浸液管媒体(BHI)和甘油为未来培养。剩下的唾液是55分成uL整除并存储在无菌cryovials。 Both BHI and saliva samples were then immediately stored at −80 °C until further processing.
146名参与者,三受试者自愿退出七并不包括在这项研究由于唾液分泌不足(< 55 uL) 136份唾液样本。样品采集后,研究参与者的电子医疗记录协调员访问病人诊断信息包含在小说主题ID号。诊断是用来确定健康状况和评估疾病的阶段当每个样本。各种诊断被分成以下类别:胰腺癌,其他疾病(包括胰腺疾病、non-pancreatic消化疾病/癌症和非消化道疾病/癌症),和健康(十几)控制。健康个体被定义为参与者没有记录慢性消化或非消化道疾病,和5年解决任何先前记录的消化或非消化道疾病。排除标准包括参与者接受积极的化疗或放射治疗或使用抗生素前两周唾液收集以及侵入性手术在过去的一年。
隔离DNA, PCR和16 s rRNA测序
细菌DNA从50 uL的患者唾液中直接提取使用该款PowerSoil DNA提取工具包(目录12888 - 05年,莫生物实验室,卡尔斯巴德,CA,美国制造商的协议。基因组DNA是量化使用NanoDrop™分光光度计和存储−20°C。
16 s rRNA (rRNA)扩增子区域使用条形码放大“普世”细菌引物515 f (5′-AATGATACGGCGACCACCGAGATCTACAC TATGGTAATT GT GTGCCAGCMGCCGCGGTAA-3′)和806 r (5′-CAAGCAGAAGACGGCATACGAGAT XXXXXXXXXXXX AGTCAGTCAG CC GGACTACHVGGGTWTCTAAT-3′) (X表示12个基点的位置条码)与Illumina公司适配器使用的地球微生物项目(http://www.earthmicrobiome.org/emp-standard-protocols/16s)。条形码引物允许池的多重PCR扩增子在一个序列运行。PCR反应条件进行了使用了地球微生物工程。热循环参数如下:94°C 3分钟(变性)其次是放大了35周期在94°C 45 s, 50°C的60年代和90年代,72°C的72°C和最后一个扩展10分钟(Caporaso et al ., 2011)。PCR扩增子被测序的Illumina公司MiSeq平台核心阿贡国家实验室测序设施(Lemont, IL)。
序列分析
16 s rRNA序列de-multiplexed使用定量了解微生物生态学(QIIME v.1.8.0,http://www.qiime.org)管道。序列分为操作分类单位97%(辣子鸡)序列相似性使用绿色煤电引用数据库。辣子鸡,没有集群与已知的分类群在97%的身份数据库集群或更高新创(UCLUST (埃德加,2010)。代表每个OTU序列然后使用PyNast对齐(Caporaso, bitting &布什曼,2010),并分类被分配使用RDP分类器(2.2版)(科尔et al ., 2003)。系统发育树构建使用FastTree (价格,Dehal &阿金,2009)。执行下游分析之前,病人样本稀薄100000序列样本,单件和辣子鸡出现在< 25%的样本稀疏之前被移除。嵌合序列被确定使用ChimeraSlayer QIIME,以及解读(赖特,Yilmaz &格拉,2012),随后删除。使用QIIMEα多样性指标也进行了计算。β多样性样本之间的距离(加权和不加权的UniFrac)也进行了计算,并用来占类群的相对多度的差异和种系发生(Vazquez-Baeza et al ., 2013)。β多样性比较使用分析相似之处(ANOSIM)。我们还测试了是否有显著差异的比率与GraphPad棱镜的不同类别之间的特定属6.0版本利用克鲁斯卡尔-沃利斯检验其次是邓恩的多重比较修正。统计学意义是接受的p< 0.05。分析和识别潜在的污染物是使用SourceTracker (骑士et al ., 2011)。
定量PCR (qPCR)
纤毛菌属使用qPCR丰度决定。简单地说,对于每一个样品我们估计纤毛菌属大量使用纤毛菌属特定的16 s引物和归一化值总体细菌丰度估计用qPCR普遍细菌16 s引物(5′-TCCTACGGGAGGCAGCAGT-3′底漆,和5′-GGACTACCAGGGTATCTAATCCTGTT-3′反向引物)开发的Nadkarni et al。(2002)。qPCR上执行一个Bio-Rad CFX96触摸™实时PCR检测仪器。的最大Ct(通用阈值周期)16 s引物将35周期和Ct高于此阈值的水平被认为是背景噪音。Genus-specific引物的扩增纤毛菌属设计使用RDP 16 s rRNA序列得到分类器(2.2版)(科尔et al ., 2003)。Primer3在线软件是用于引物选择,条件解决的建议桑顿和巴苏(2011)。的纤毛菌属正向引物序列(5′-GGAGCAAACAGGATTAGATACCC-3′)和纤毛菌属反向引物序列(5′-TTCGGCACAGACACTCTTCAT-3′)生成87个基点的扩增子。PCR反应包含1嗯正向和反向纤毛菌属引物以50°C的热循环参数为2分钟,10分钟95°C和40 95°C的周期为1分钟15秒和62.5°C。通用引物,扩增反应纤毛菌属引物进行了至少重复使用25 uL SYBR绿色主人混合(Bio-Rad)和0.85 ng / uL提取的DNA作为模板。各种网络工具,包括在网上PCR扩增(Bikandi et al ., 2004)和核糖体数据库项目(科尔et al ., 2003)被用来检查的特异性寡核苷酸引物序列为目标生物。唾液样本测序(伊顿生物科学,圣地亚哥,CA)使用我们的小说引物,引物特异性进一步证实16 s rRNA BlastN搜索数据库。
结果
唾液微生物多样性配置文件生成共有108名患者。8例患者诊断为胰腺癌(P), 78年被诊断出患有其他疾病(包括癌症)(O)和22被认为是健康的(十几)控制(H)。表1细节的个人临床特征,包括性别和种族。在胰腺癌患者的108患者中,23,消化和非消化道疾病类别被诊断出患有癌症。表2细节的癌症类型,以及类别分组和癌症患者的平均年龄在每个类别。
| 人口统计资料 | 胰腺癌(P) | 其他疾病(O) | 健康的控制(H) | 总 |
|---|---|---|---|---|
| n= 8 | n= 78 | n= 22 | n= 108 | |
| 性 | ||||
| 男性 | 6 | 38 | 12 | 56 |
| 女 | 2 | 40 | 10 | 52 |
| 种族 | ||||
| 高加索人 | 6 | 56 | 15 | 77年 |
| 拉美裔 | 2 | 6 | 5 | 13 |
| 亚洲 | 0 | 4 | 1 | 5 |
| 未知的 | 0 | 12 | 1 | 13 |
| 癌症的分类 | 年龄的意思 | N |
|---|---|---|
| 胰腺(n= 8) | 71.1 | |
| 胰腺癌 | 8 | |
| 消化系统(n= 9) | 64.7 | |
| 壶腹 | 3 | |
| 食管 | 3 | |
| 胃 | 1 | |
| 直肠 | 2 | |
| 非消化道(n= 6) | 54.8 | |
| 乳房 | 1 | |
| 皮肤 | 1 | |
| 睾丸 | 1 | |
| 甲状腺 | 3 |
Illumina公司测序产生大约680万在所有样本序列。上可用的序列是FigShare (http://dx.doi.org/10.6084/m9.figshare.1422174)随着映射文件(http://dx.doi.org/10.6084/m9.figshare.1422175)。分析潜在的示例使用SourceTracker污染(骑士et al ., 2011)发现一些人类皮肤和/或环境污染的证据。相关的序列辣子鸡确认为污染物,主要是葡萄球菌(皮肤)和蓝藻(叶绿体),被从所有后续分析。从这些数据中,我们发现了一个共有12个细菌类群和139属。变形菌门、放线菌、拟杆菌、厚壁菌门和Fusobacteria 5个主要类群,占99.3%的口腔细菌(图1)。变形菌门的平均相对丰度较低的胰腺癌患者相对于其他样本类别,而厚壁菌门往往更高,尽管这些没有重大调整后为多个比较(罗斯福)。胰腺癌组也有更高水平的纤毛菌属以及低水平的Porphyromonas,奈瑟氏菌属(图2)。一般来说,多级分类的健康组类似于其他的疾病组,而胰腺癌组distinctive (无花果S1。)。然而,没有明显差异的三个主要组(H、O和P)β多样性(ANOSIM;P= 0.1)或α多样性(Chao1,知道测试;P= 0.6;信仰的PD,知道测试;P= 0.56)。
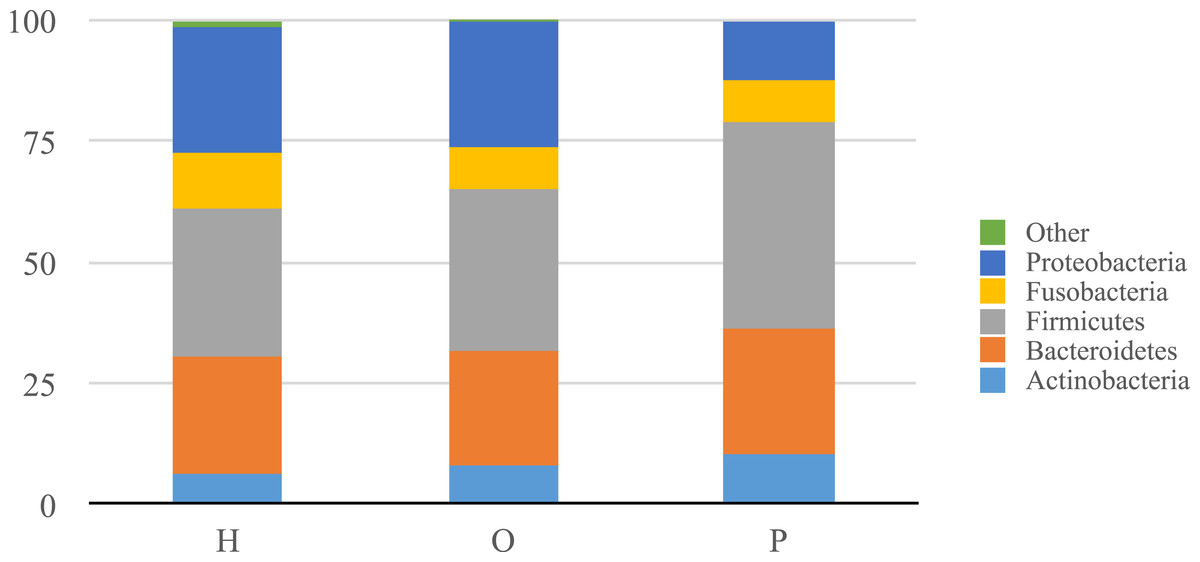
图1:相对丰富的类群中确定病人唾液诊断小组总结。
相对丰富的类群在口头总结患者的社区从108年开始研究诊断组(H,健康的控制;啊,其他疾病;和P,胰腺癌)。{kind=link}
在先前的研究法雷尔et al。(2012)和林et al。(2013),我们看到的相对丰度较低奈瑟氏菌属和Aggregatibacter,虽然这些差异不显著(知道测试;P= 0.07,P= 0.09)。细菌型更丰富的胰腺癌患者与健康人相比,类似林等人观察,尽管这也不显著(知道测试;P= 0.27)。我们没有看到任何的相对丰度的差异链球菌或Granulicatella之前,显示不同的胰腺癌研究(法雷尔et al ., 2012)。额外的分析目标是基于我们的第一个61份唾液样本组成的初步研究(包括3胰腺癌患者),显示显著更高纤毛菌属和更低的Porphyromonas在胰腺癌患者的唾液。
的丰度比纤毛菌属,特别是两个辣子鸡(任意命名OTU 31235和OTU 4443207)Porphyromonas在胰腺癌患者明显高于(图3)。一个爆炸的比较这些辣子鸡16 s序列在人类口腔微生物数据库(陈et al ., 2010)(HOMD RefSeq版本13.2)发现OTU 31235 100%相似纤毛菌属sp。口服分类单元221,虽然OTU 4443207类似于99.3%纤毛菌属hongkongensis。我们发现一个强大的正相关(皮尔森的相关性r= 0.903,P= 0.0000001)之间纤毛菌属丰度获得16 s rRNA测序(OTU相对丰度)和实时qPCR (图4)。
讨论
我们的唾液微生物分析概要支持之前的工作表明唾液微生物群落的患者被诊断出患有胰腺癌从唾液微生物群落的健康的患者或其他疾病的患者,包括non-pancreatic癌症。在门级,胰腺癌患者体内壁厚菌门的微生物的比例相对较高和较低的比例变形菌门(图1)。在细分类水平,我们观察到特定的平均相对丰度差异属胰腺癌患者与其他患者组(图2)。例如,有一个更高的比例纤毛菌属在胰腺癌患者中,同时的比例Porphyromonas和奈瑟氏菌属在这些患者低。
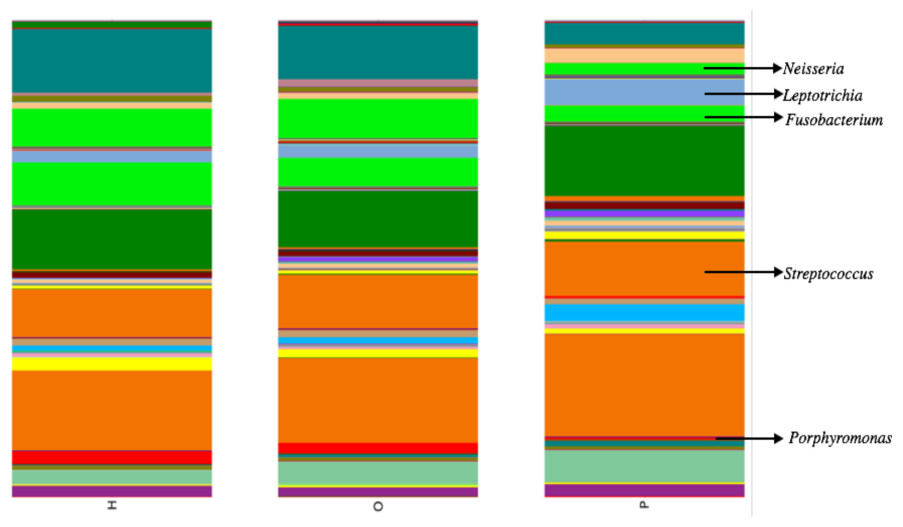
图2:意味着相对丰度相比,胰腺癌患者(P)特定属健康(H)和其他疾病(O)患者组。
属的相对丰度从108例口服社区。箭头指向特定的跨诊断组属显示有趣的趋势。{kind=link}
之间最显著的区别我们发现微生物的胰腺癌的病人和其他病人群体在细菌属的比例纤毛菌属和Porphyromonas(LP比率)(图3)。LP比率被确定为潜在生物标志物的初步分析和完整的数据集的分析发现明显高于胰腺癌患者唾液的LP比率比其他病人群体。使用另一种方法来验证这些差异,我们旨在的相对丰度纤毛菌属(图4)。有趣的是,在16 s rRNA的分析数据,我们成功地使用了LP比例重新分类的病人之一non-pancreatic癌症疾病组。这个特定的个体最初被诊断为有一个未知的消化系统疾病,但病人的高LP比率表明胰腺癌(图3)。随后,病人重新评估和诊断出患有胰腺癌,支持了LP比率可能作为胰腺癌生物标志物。

图3:的相对丰度纤毛菌属来Porphyromonas不同病人之间的类别。
每个符号代表的比例纤毛菌属口服分类单元221和纤毛菌属hongkongenesis来Porphyromonas个别病人(n= 108)。患者分为3种不同的类别根据其诊断:健康控制(H)、其他疾病(包括癌症)(O),和胰腺癌(P),单杠和误差代表均值和SEM,分别。∗∗∗p< 0.001(克鲁斯卡尔-沃利斯检验其次是邓恩的多重比较检验)。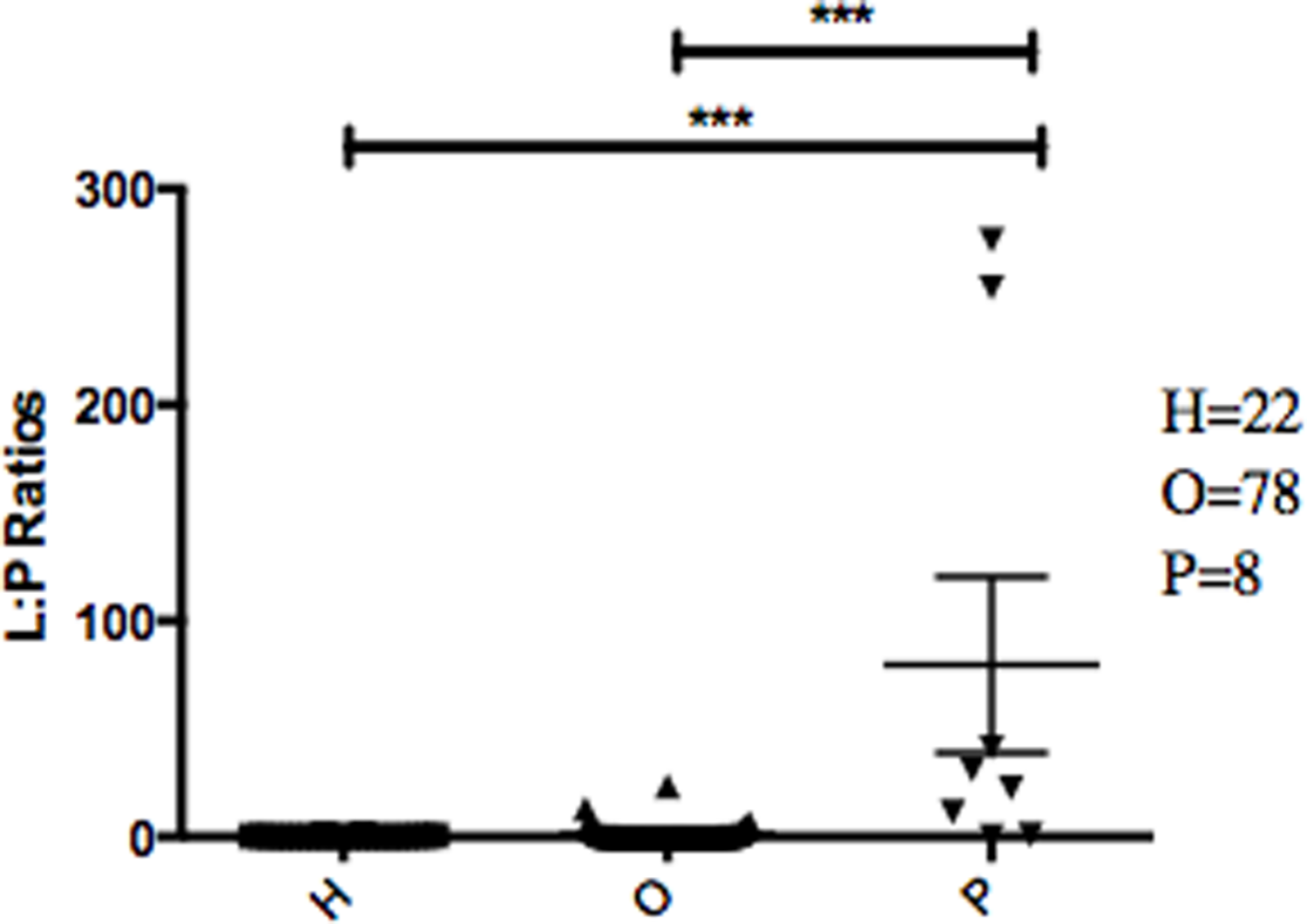{kind=link}
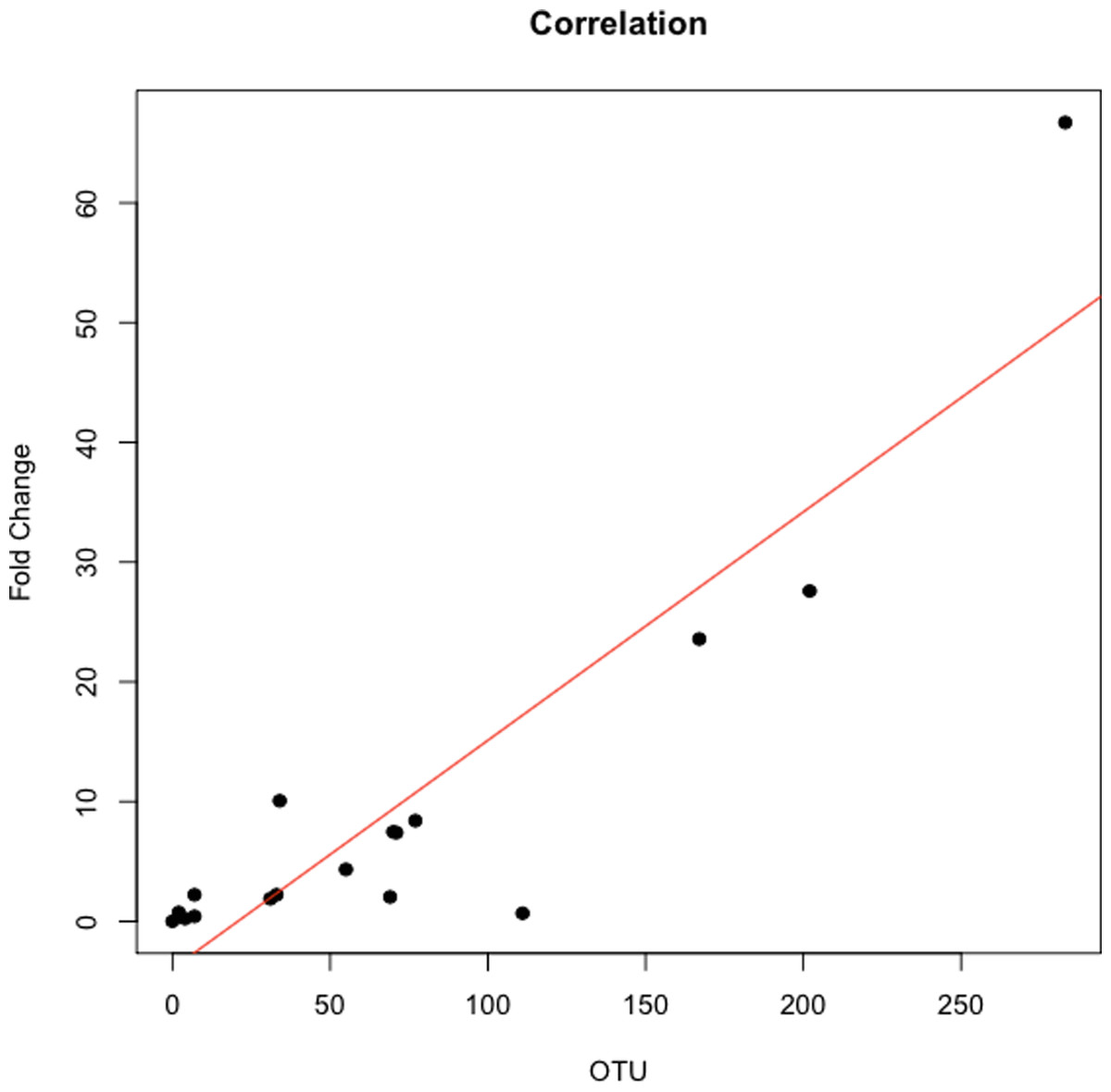
图4:之间的相关性纤毛菌属数量从16 s rRNA序列和实时qPCR。
交叉验证的总纤毛菌属使用实时qPCR OTU丰富。在使用16 s rRNA作为参考基因的水平正常化纤毛菌属属、数据规范化的褶皱变化三个健康对照组相对较低纤毛菌属OTU丰富。每个符号代表一个病人:P = 6, O = 12。纤毛菌属OTU丰度是与qPCR褶皱变化根据皮尔逊相关性(r= 0.903)。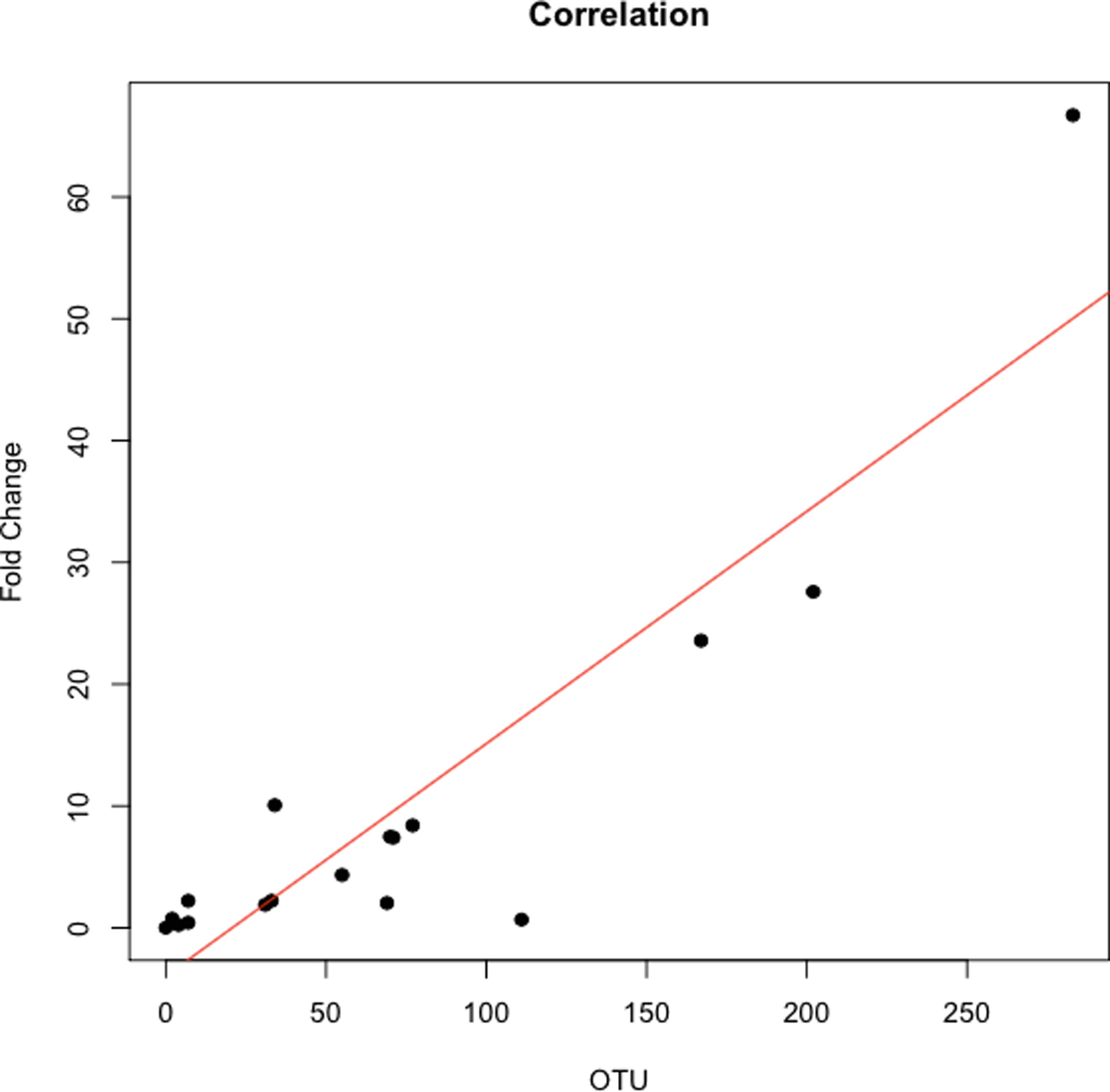{kind=link}
尽管小患者群在这项研究中,我们相信我们的结果尤其值得注意,因为我们能够区分胰腺癌患者和各种其他疾病患者(包括non-pancreatic癌症),除了健康的控制。其他研究者提出的使用比率的细菌类群。Galimanas et al。(2014)建议使用唾液细菌丰度比值作为区分意味着健康和患病的病人。分类比率被用来区分受试者在研究肥胖的(Lazarevic et al ., 2012)、糖尿病(张&张,2013)和牙周疾病(Moolya et al ., 2014)。率比较高水平的分类也有助于控制个体之间的差异(丁&城堡,2014;塞格雷,2012;Schwarzberg et al ., 2014;王et al ., 2013)。
回顾文献显示纤毛菌属在口腔健康的作用仍然是难以捉摸的。然而,这些细菌在职业病人的血液中发现了(Eribe &奥尔森,2008)和共现明显与结直肠肿瘤(沃伦et al ., 2013)。纤毛菌属被孤立于心血管和胃肠道脓肿,从系统性感染,和被认为是致病性(汉&王,2013)。关于Porphyromonas抗体,Porphyromonas gingivalis一直在与胰腺癌(直接相关米肖德et al ., 2012)。欧洲队列研究测量等离子体抗体25口腔细菌pre-diagnostic血液样本405个胰腺癌病人和416个匹配控制和发现倍增加胰腺癌的风险在那些高致病性毒株的抗体滴度p . gingivalis(米肖德et al ., 2012)。乍一看,似乎矛盾的患者更高Porphyromonas抗体滴度将口腔丰度较低。然而,研究特定periopathogens包括系统性免疫的动物Porphyromonas显示了殖民的口腔细菌和减少牙周炎(埃文斯et al ., 1992;佩尔松et al ., 1994;克拉克et al ., 1991)。同样,高Porphyromonas抗体滴度在个人与胰腺癌可能会降低他们的口头丰富,尽管这种联系需要正式测试。
唾液微生物多样性的变化也可能是一个系统对胰腺癌的回应。胰腺癌是削弱免疫系统(冯Bernstorff et al ., 2001),这可能导致口腔细菌过度生长和转向系统入侵牙周病原体。细菌病原体的扩散可以帮助癌症恶化通过系统性炎症(El-Shinnawi & Soory, 2013)或免疫干扰(Feurino,张&巴拉,2007)。因此,最初的增加Porphyromonas可能是紧随其后的是减少由于系统入侵和抗体生产。事实上,炎症被认为扮演了一个重要的角色在胰腺癌的发展(法罗&埃弗斯,2002)。
我们也比较了相对丰度表示的其他细菌属作为潜在生物标志物在以前的工作法雷尔et al。(2012)。像法雷尔et al .,我们发现的比例较低奈瑟氏菌属在胰腺癌患者唾液与健康和其它疾病类别相比,尽管这一趋势不显著。然而,我们没有发现相同的结果作为其他细菌属法雷尔等人。我们的数据也显示增加拟杆菌和减少大量的细菌属Aggregatibacter在胰腺癌患者,支持一项初步研究的结果林et al。(2013),但无论是趋势显著。
我们的研究方法论的差异尤其是法雷尔等人的研究,可能部分解释我们的结果的不同。例如,不能the16S V4地区的核糖体rna基因歧视链球菌从其他链球菌物种可能阻止我们检测的区别在这个物种的丰度(法雷尔et al ., 2012)。此外,我们的研究有一个广泛的类别和癌症病人并不总是局限于胰腺的抽样。
有趣的是,自从完成我们的研究中,Mitsuhashi et al。(2015)口头的检测报告梭菌属在胰腺癌组织中。我们丰富的回顾性研究数据还发现相对丰度较低梭菌属胰腺癌患者与其他患者类别(图2;知道测试,P罗斯福校正前= 0.03)表明流程驱动的差异梭菌属可能类似于我们提出的机制Porphyromonas。虽然结果不显著调整后的多重比较(罗斯福),我们建议梭菌属丰度应考虑作为一个潜在的生物标记目标未来的研究与更大的患者群。
总体而言,我们的研究表明,唾液微生物组的成员承诺作为潜在胰腺癌生物标志物,我们可能发现了一个重要的在这方面(即新局面。LP比率)。然而,我们相对较少的胰腺癌患者和样本之间的差异,我们的发现和以前的工作表明,需要更大的患者群,以确定唾液诊断有用的生物标志物。未来的研究应该关注改进元数据集合,包括饮食和口腔健康信息(即。,periodontal disease), which would make it possible to run statistical analyses that control for multiple factors involved in shaping oral microbial diversity. It will also be important to sample the same individual’s saliva over time to assess whether we can distinguish between disease stages and also to control for intra-individual variation. Further, it is possible that single biomarkers may never be able to consistently identify pancreatic patients from other conditions. Thus, we may need more complex metrics that combine the abundances of multiple salivary bacteria, metabolite profiles, and detailed patient metadata. Effective diagnostic biomarkers for pancreatic cancers have been difficult to find, but are sorely needed and have the potential to save thousands of lives each year.
补充信息
多级分类的病人唾液诊断小组总结
相对丰度(A)类,(B), (C)的家庭,(D)属口服社区从108年研究的患者根据诊断。病人被诊断分组(H,健康;啊,其他;P,胰腺癌)。