条文本

摘要
背景最近的证据表明,微生物组在胰腺导管腺癌(PDAC)的病因和进展中的作用。
客观的探讨粪便和唾液菌群作为潜在的诊断生物标志物。
方法我们将霰弹枪宏基因组和16S rRNA扩增子测序应用于来自西班牙病例对照研究(n=136)的样本,包括57例慢性胰腺炎患者,50例对照组和29例处于发现期的慢性胰腺炎患者,以及来自德国病例对照研究(n=76)处于验证期的样本。
结果粪便宏基因组分类器的表现比基于唾液的分类器要好得多,基于一组27种微生物种类,识别PDAC患者的接受者工作特征曲线(AUROC)下面积的准确性高达0.84,在疾病早期和晚期都具有一致的准确性。当我们将基于微生物的预测与血清碳水化合物抗原(CA) 19-9水平结合在一起时,性能进一步提高到0.94 AUROC, CA 19-9是目前唯一的非侵入性、食品和药物管理局批准的低特异性PDAC诊断生物标志物。此外,基于微生物的分类模型仅限于富含pdac的物种,在针对25个公开的宏基因组研究人群(n=5792)的各种健康状况进行验证时,具有高度的疾病特异性。两种基于微生物的模型在德国验证人群(n=76)上具有较高的预测精度。通过16S rRNA测序和荧光原位杂交,在胰腺肿瘤和非肿瘤组织中检测到几种粪便PDAC标记物种。
结论总之,我们的结果表明,基于粪便微生物的非侵入性、健壮性和特异性筛选用于早期检测PDAC是可行的。
- 胰腺癌
- 肠道微生物
- 癌症预防
- 胰腺肿瘤
- 筛选
数据可用性声明
数据可在一个公共的、开放访问的存储库中获得。所有与研究相关的数据都包含在文章中或作为补充信息上传。样本的原始测序数据可在欧洲核苷酸档案(ENA)中获得,研究标识号为PRJEB38625和PRJEB42013。这些示例的元数据见补充表S1和S2。作为统计建模管道输入的过滤的分类和功能概要可在补充数据S1和S2中获得。分析代码和结果可在https://github.com/psecekartal/PDAC.git.
这是一篇开放获取的文章,按照创作共用署名4.0未移植(CC BY 4.0)许可发布,该许可允许其他人复制、重新发布、混合、转换和基于此作品的任何目的,只要原始作品被正确引用,提供许可证链接,并说明是否进行了更改。看到的:https://creativecommons.org/licenses/by/4.0/.
统计数据来自Altmetric.com
本研究的意义
关于这个主题我们已经知道了什么?
胰导管腺癌(PDAC)在全球范围内呈上升趋势,造成了很高的疾病负担和死亡率,但仍然没有准确的、非侵入性的诊断选择。
口腔、粪便和胰腺微生物组组成的改变与PDAC风险的增加有关。
新的发现是什么?
以粪便微生物为基础的分类器被描述为预测PDAC具有高准确性和特异性,独立于疾病阶段,具有作为非侵入性诊断试剂的潜力。
在西班牙的一个队列中,基于27个物种,粪便宏基因组分类器在接受者工作特征曲线(AUROC)下识别PDAC的面积精度为0.84。当与特异性较低的碳水化合物抗原(CA) 19-9血清标志物结合时,准确性提高到0.94 AUROC。
分类器在一个独立的德国PDAC队列(0.83 AUROC)中得到验证,PDAC疾病特异性在25个具有各种健康状况的公开可得宏基因组研究人群中得到证实(n=5792)。
粪便样本中富集的标记类群(韦永氏球菌属,链球菌,Akkermansia)以及健康和肿瘤胰腺组织中丰度差异的分类群(拟杆菌,乳酸菌,双歧杆菌属)通过荧光验证原位杂交。
本研究的意义
在可预见的未来,它会对临床实践产生怎样的影响?
基于粪便微生物的PDAC检测可能为PDAC的早期诊断提供一种无创的、经济的和稳健的方法。
提出的pdac特异性微生物组特征,包括跨组织微生物群之间的联系,为疾病病因、预防和可能的治疗干预提供了新的微生物相关假设。
简介
胰腺导管腺癌(PDAC)是最常见的胰腺癌,也是癌症相关死亡的主要原因,尽管发病率相对较低。1 2PDAC的高致死率是诊断晚和治疗选择有限的结果3.:症状无特异性,通常只在疾病晚期出现,此时肿瘤可能局部不可切除,或表现为转移性疾病。目前,PDAC的诊断是通过影像学检查。4因此,早期检测PDAC的敏感和负担得起的检测可以改善结果。已经在胰腺组织中探索了PDAC标记,5尿液6 7和血清。8 9迄今为止,唯一的食品和药物管理局(FDA)批准的PDAC生物标志物仍然是血清碳水化合物抗原(CA) 19-9。CA19-9具有有限的疾病特异性,因为在其他几种伴随情况下(如胆道梗阻),其水平可能升高,因此主要用作PDAC监测的标记物,而不是筛查或诊断。10 - 14
PDAC的病因复杂,已知的危险因素包括年龄、慢性胰腺炎、糖尿病、肥胖、哮喘、血型和生活方式(如吸烟和大量饮酒)。15日16这些危险因素在PDAC病因中的作用也可能是由微生物组的改变所补充的,有时甚至是介导的。例如,糟糕的口腔卫生和牙周炎与PDAC风险的增加有关,17这一观察结果也延伸到牙周炎和龋齿相关的微生物物种。18 19这些物种的变化有时是口腔微生物群中更广泛的组成变化的一部分20 21或作为PDAC的风险因素本身进行了研究。22同样,肠道中的微生物组成第23 - 25和十二指肠,26日27日通过16S rRNA扩增子测序进行量化,之前已经与PDAC风险有关。
人类的胰腺拥有一种与口腔和肠道共享物种的微生物群,25 28-32尽管它的确切组成仍然难以捉摸,因为在细菌生物量低的样品中与污染控制相关的挑战。33在小鼠模型中,来自肠道的微生物可以促进胰管的癌变,25 30这表明微生物组在PDAC病因和进展中的作用,最近扩展到真菌。34此外,胰腺肿瘤微生物组也可能与PDAC患者的疾病进展和长期生存有关。31
然而,将这些进展转化为用于临床应用的pdac特异性微生物组特征,到目前为止,在很大程度上仍未探索。在这里,我们基于对西班牙(ES)研究人群的宏基因组调查,对57例新诊断的PDAC患者和treatment-naïve例PDAC患者、29例慢性胰腺炎(CP)患者和50例匹配对照进行了鉴定。我们对唾液、粪便、胰腺正常组织和肿瘤组织进行了采样,并使用全基因组鸟枪宏基因组学、16S rRNA扩增子测序和荧光技术评估了微生物组成原位杂交(FISH)分析。PDAC患者和非PDAC受试者之间的最佳区分是通过基于一组27种粪便微生物的统计模型实现的,这些粪便微生物可以在诊断设置中以有针对性的方式量化。在一个独立的德国(DE) PDAC验证人群中,包括44名PDAC患者和32名对照组,证实了基于微生物的模型的预测准确性,并在结合血清CA19-9水平时进一步提高。根据来自9种疾病的25项研究(n=5792)的现有数据,我们进一步验证了这些模型的疾病特异性。35-59在癌症组织中也检测到一些富含PDAC的物种,它们可能与口腔和肠道种群有关,如前所述,支持它们在PDAC发病机制中的潜在作用。25 30 31 34
方法
受试者招募和样本收集
采用病例对照设计。2016年至2019年期间从马德里Ramón y Cajal医院和西班牙巴塞罗那Vall d 'Hebron医院前瞻性招募受试者,使用相同的生物样本收集、处理和存储协议。新诊断为PDAC的受试者(n=57),年龄为18岁,在接受任何癌症治疗前被确诊。在疑似PDAC的受试者中进行招募,并在任何治疗前进行采样。慢性胰腺炎患者(CP, n=29)来自同一家医院。从与PDAC危险因素无关的住院患者中选择符合年龄、性别和医院的对照。排除因身体能力障碍而不能参与研究的参与者。机构审查委员会伦理批准(CEI PI 26 2015-v7)和书面知情同意分别来自参与中心和研究参与者。流行病学和生活方式数据由训练有素的监测人员通过结构化问卷在面对面访谈中收集。临床数据,包括疾病的阶段和随访数据,由相同的监测人员从医院图表中检索,同样使用结构化问卷。 Recorded jaundice status was additionally confirmed and extended by direct bilirubin measurements from blood samples in CNIO, Madrid. All data were entered, edited and managed using REDCap. Missing lifestyle and medication values in the metadata (missing overall in 3.1%) were imputed using a random forest-based algorithm for missing data imputation called missForest (n=100 trees).60根据imputation误差估计,imputation精度较高(平均袋外误差=0.12)。根据德国马尔堡实验室医学和病理生物化学研究所的制造商说明,采用电化学发光免疫分析法(ECLIA,德国罗氏诊断公司)分析血清CA19-9水平。每个样品重复检测,阳性对照检测在每个培养皿中(在线补充表S1).
粪便和唾液(漱口水)样本保存在RNALater中,在4℃下立即保存12小时,然后转移到−20℃下再保存24小时,然后在−80℃保存直到DNA提取。在手术中收集了一部分个体的肿瘤和未受影响的组织样本,在病理评估后立即在液氮中快速冷冻,并在−80°C保存。所有的样品都是用干冰运输的。
在埃尔兰根大学医院外科(32个PDAC和32个对照样本)和法兰克福歌德大学临床内科I部转化肝脏科(12个PDAC样本)招募了一个独立的验证人群,使用相同的生物样本收集、处理和存储协议。匹配对照从入院时的初步诊断与PDAC危险因素无关的住院患者中选择。该研究获得了当地伦理委员会(SGI-3-2019, 451_18 B)的批准,并获得了研究参与者的书面知情同意。临床资料,包括疾病分期及随访资料,是从有关病人的医院图表的临床记录(在线补充表S2).血清CA19-9水平分析采用常规免疫分析法(德国罗氏诊断公司),按照制造商的说明。粪便样本保存在OMNIgene-Gut OM-200小瓶中(Steinbrenner Laborsysteme GmbH, Germany),并立即保存在−80°C,直到DNA提取。
样品处理
粪便和唾液样本在冰上解冻,并根据制造商的说明使用Qiagen Allprep PowerFecal DNA/RNA试剂盒提取基因组DNA (Qiagen, Hilden, Germany)。从胰腺肿瘤和非肿瘤组织样本中提取基因组DNA,使用Qiagen DNeasy血液和组织试剂盒,该试剂盒采用Del Castillo改进方案等26:机械裂解细胞(用5mm不锈钢珠,25 Hz, 150 s),然后溶菌酶处理(20 mg/mL),蛋白酶和RNAse消化(56°C, 2 h)。所有样品被随机分配到提取批次。为了考虑提取、聚合酶链反应(PCR)和测序试剂盒的潜在细菌污染,我们在每个组织DNA提取批次(在线补充图1).
16S rRNA扩增子测序
使用引物331F (5 ' - tcctacgggaggcagcagcat -3 ')在预扩增PCR中富集胰腺组织DNA为16S rRNA61和979R (5 ' -GGTTCTKCGCGTTGCWTC-3 ')。62循环条件包括模板在98°C初始变性2分钟,然后在98°C变性10秒,65°C退火20秒,72°C延伸30秒,72°C最终延伸10分钟。随后,使用SPRIselect磁珠(0.8左尺寸;贝克曼库尔特,布雷亚,加利福尼亚州,美国)。粪便和唾液DNA未被预先扩增。
靶向扩增16S rRNA V4区(引物序列F515 5 ' - gtgccagcmgccgggtaa -3 '和R806 5 ' -GGACTACHVGGGTWTCTAAT-3 '),63采用KAPA HiFi HotStart PCR混合试剂盒(Roche, Basel, Switzerland),采用两步条形码PCR协议(NEXTflex 16S V4 Amplicon-Seq Kit;生物科学公司,奥斯汀,德克萨斯州,美国)根据制造商的说明进行了微小的修改。聚合产物使用SPRIselect磁珠(左尺寸0.8)进行纯化,然后在海德堡欧洲分子生物学实验室基因组学核心设施的Illumina MiSeq (Illumina, San Diego, California, USA)上以2×250 bp进行测序。
16S rRNA扩增子数据处理
使用DADA2对原始读数进行质量修剪、去噪和过滤以对抗嵌合PCR产物。64使用MAPseq对得到的扩增子序列变体(ASVs)进行分类,并以98%的序列相似性映射到操作分类单元(OTUs)的参考集。65使用Infernal将不能准确映射到参考文献的Reads对准细菌和古菌二级结构感知小亚基rRNA模型66用hppc - clust聚类成平均连锁98%的otu,67如前所述。68结果,我们获得了两种分辨率的分类表:100%相同的asv和98%开放参考OTUs;除另有说明外,正文中的分析均指OTUs。
计数表通过去除保留在500个reads以下的样本和在少于5个样本中观察到的类群来过滤噪声;这从数据集中删除了2.5%的总读取。对于18个唾液样本,在确认它们与群落组成密切相关后,进行技术重复合并。对于胰腺组织和肿瘤样本,在阴性对照样本中观察到的ASVs被移除,已知试剂试剂盒污染物的读数也被移除。33在这些步骤之后,我们从143个受试者中保留了308个16S rRNA扩增子样本用于进一步分析(130个唾液,118个粪便,20个未受影响的胰腺组织,23个肿瘤组织和17个匹配的PDAC组织样本)。
鸟枪宏基因组测序
根据起始材料的浓度,使用NEB Ultra II和SPRI HD试剂盒制备212份粪便和100份唾液样本的宏基因组文库,目标插入尺寸为350,并在2×150 bp的配对端设置的Illumina HiSeq 4000平台(Illumina, San Diego, California, USA)上测序,每个样本的目标深度为8 Gbp,位于海德堡欧洲分子生物学实验室基因组学核心设施。每个样本的排序统计信息在相关的git存储库中提供(https://github.com/psecekartal/PDAC.git).对于三个唾液样本和一个粪便样本,在确认它们与群落组成密切相关后,进行技术重复合并。
宏基因组数据处理
宏基因组数据使用NGLess v0.7.1中建立的工作流程进行处理。69对原始reads进行质量修剪(Phred评分≥25时≥45 bp),并根据人类基因组(hg19版本,≥45 bp时映射≥90%的一致性)进行过滤。将过滤后的reads与从proGenomes数据库v2中获得的5306个物种级基因组簇的代表基因组进行映射(≥97%的一致性跨越≥45 bp)。70
使用mOTU剖面仪v2.5获得分类学剖面71并过滤,只保留相对丰度≥10的观察到的物种−5≥2%的样品。基因功能图谱是从全球微生物基因目录(GMGCv1, Coelho等72,http://gmgc.embl.de/),通过从eggNOG v4.5中总结读取计数73对同源组和KEGG模块的注释。相对丰度≥10的特征−5≥15%的样品留作进一步分析。
微生物组数据统计分析
所有数据分析都在R统计计算框架v3.4或更高版本中进行。
细化的每样本类群多样性(“alpha多样性”,平均超过100次稀疏迭代)计算为Hill系数q=0(即类群丰富度)、q=1 (Shannon熵的指数)和q=2(辛普森指数的倒数)的有效类群数量,均匀度度量为其比值。除另有说明外,正文中的结果均指类群丰富度。采用方差分析(ANOVA)检验alpha多样性的差异,随后采用事后检验和Benjamini-Hochberg校正,如正文所述。
群落组成的样本间差异(“beta多样性”)被量化为原始或平方根转换计数、丰度加权Jaccard指数、丰度加权和非加权TINA指数上的布雷-柯蒂斯差异,如前所述。74除非另有说明,这些指数之间的趋势一般是一致的。结果报告的布雷-柯蒂斯不相似的非转换数据。利用PERMANOVA的“adonis2”实现和R包纯素v2.5中的基于距离的冗余分析,对群落组成与微生物-外部因素的关联进行了量化。75为了量化单个类群的丰度和学科特定变量之间可能引起混淆的单变量联系(见正文),我们根据丰度分布(在线补充图2-3而且在线补充表S4-S5).从血液样本中测定胆红素水平,并通过临床记录确认黄疸状态。由于有几个人没有黄疸状况,用于进一步分析的值是根据现有数据推算的(图1,在线补充表S1-S3).
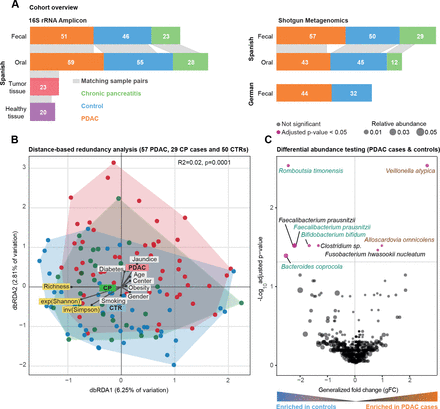
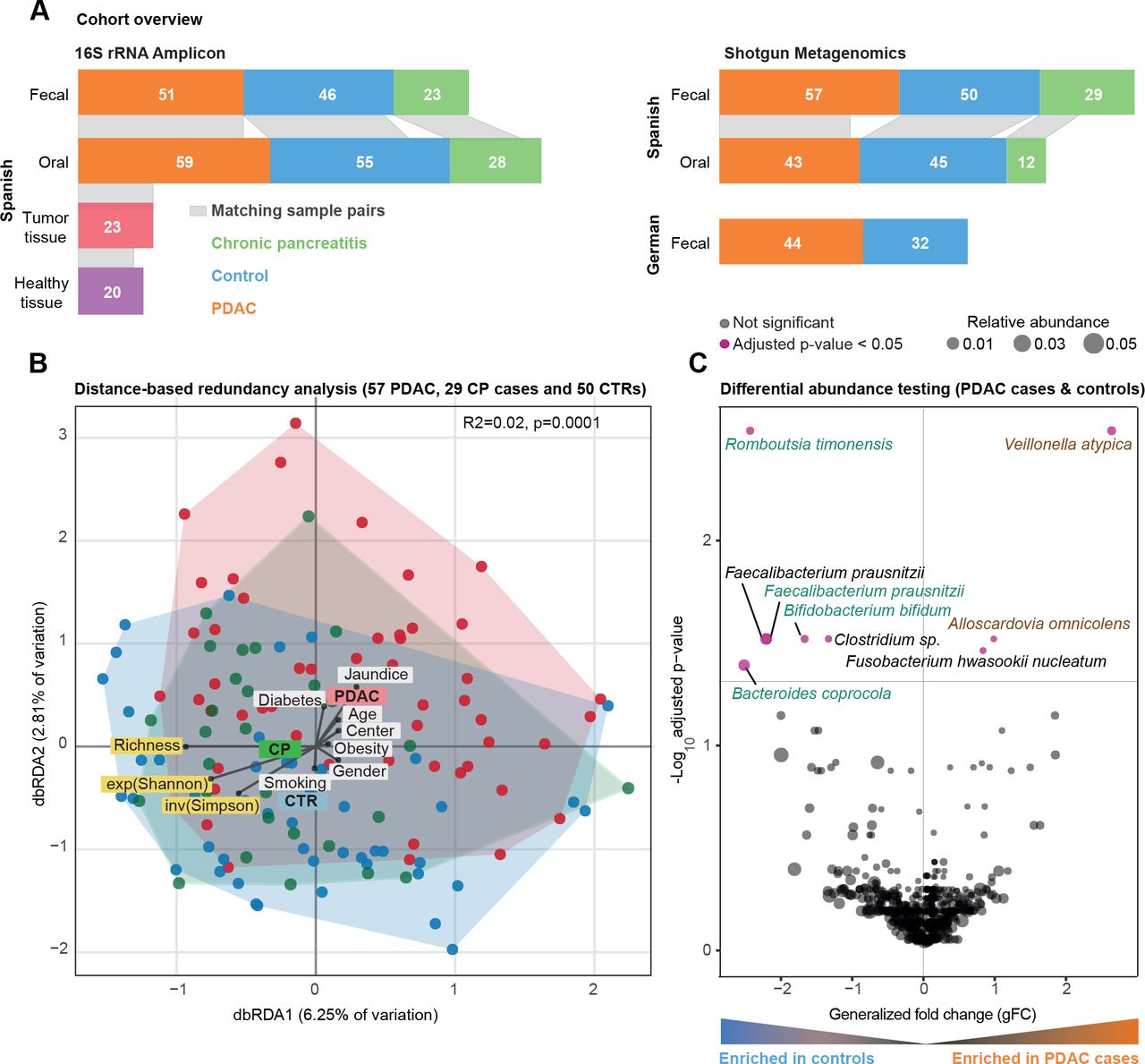
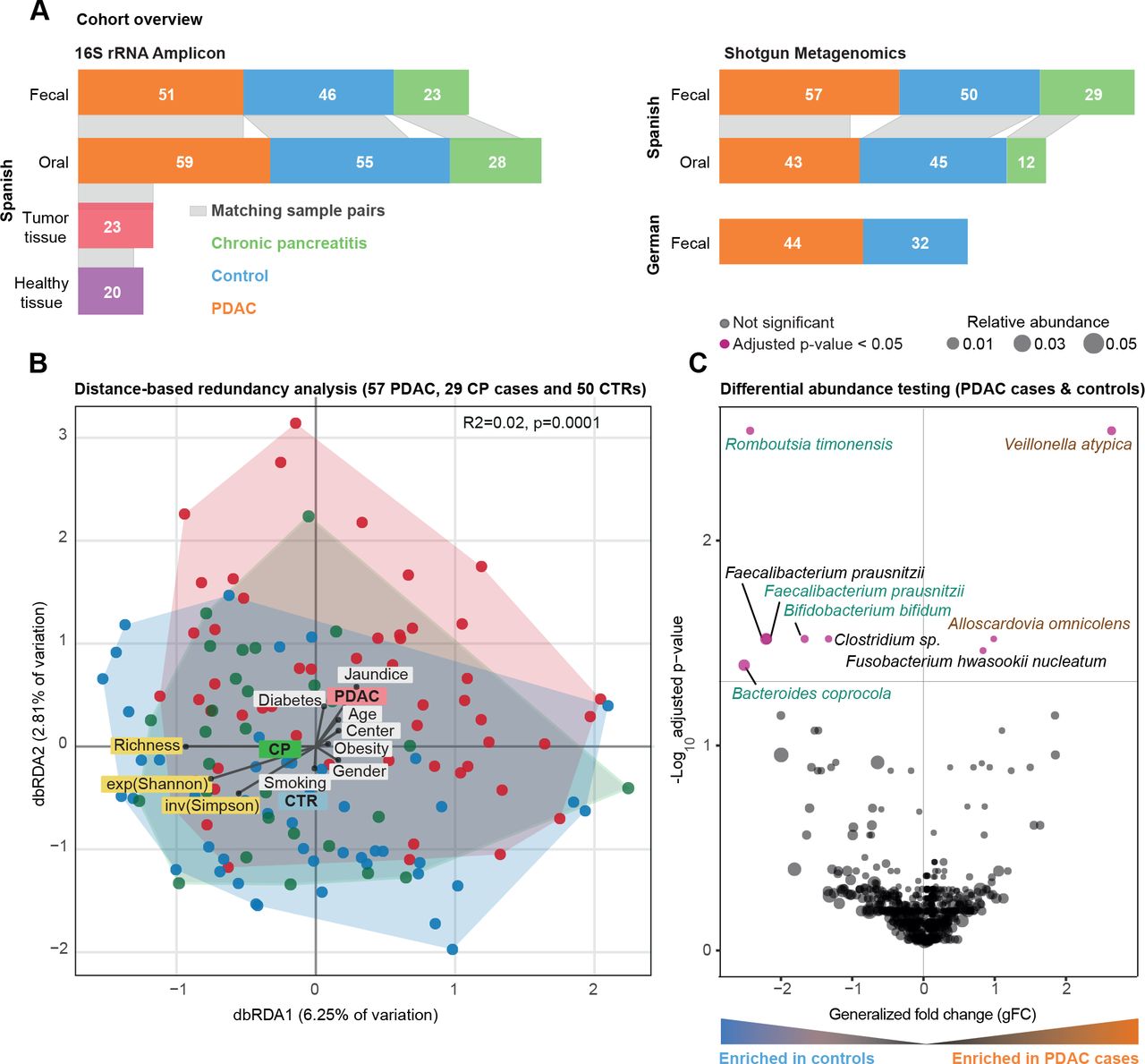
西班牙粪便微生物群数据的群落分析。(A)研究人群概况。条形图之间的灰色带表示个体内匹配的身体部位样本。(B)西班牙(ES)队列中胰导管腺癌(PDAC)、慢性胰腺炎(CP)和对照组(CTR)粪便微生物组数据的bry - curtis基于距离的冗余分析(dbRDA)。PDAC样本显示为红色圆圈,CP患者显示为绿色圆圈,对照组显示为蓝色圆圈。丰富度、指数Shannon (exp(Shannon))和反向Simpson (inv(Simpson))多样性度量也用箭头可视化,类似于测试的元数据变量。元变量与中心的距离表示混杂效应大小(见“方法”)。(C) ES粪便微生物组数据的Wilcoxon检验结果,以测试PDAC和对照病例之间富集的分类群(见“方法”)。y轴为log10(FDR校正后的p值),x轴为广义折数变化,点的大小表示给定物种的相对丰度。红点代表两组物种丰度差异显著,黑点代表FDR校正后的不显著物种。 Green and brown-coloured species are selected in metagenomic model-1 as predictors of PDAC. FDR, false discovery rate.
多变量统计建模与模型评价
为了训练预测胰腺癌的多变量统计模型,我们首先剔除总体丰度和患病率较低的类群(丰度分界点:0.001)。然后,通过log10转换将特征归一化(为了避免来自对数的无限值,在所有值中添加1e-05的伪计数),然后标准化为中心log-ratio (log.clr)。数据被随机分成测试集和训练集,进行10次重复的10倍交叉验证。对于每个测试褶皱,剩余的褶皱被用作训练数据来训练l1 -正则化(LASSO)逻辑回归模型76使用LiblineaR R包v2.10中的实现。77然后使用训练过的模型预测遗漏测试集,最后,使用所有预测计算受试者工作特征曲线(AUROC)下的面积(图2).
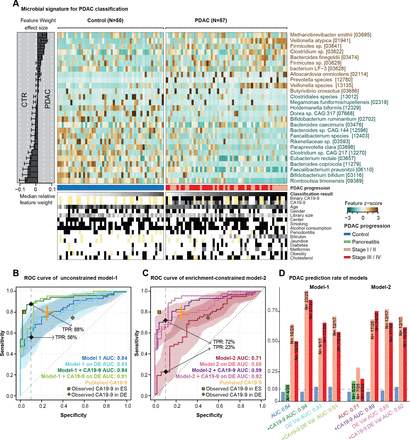
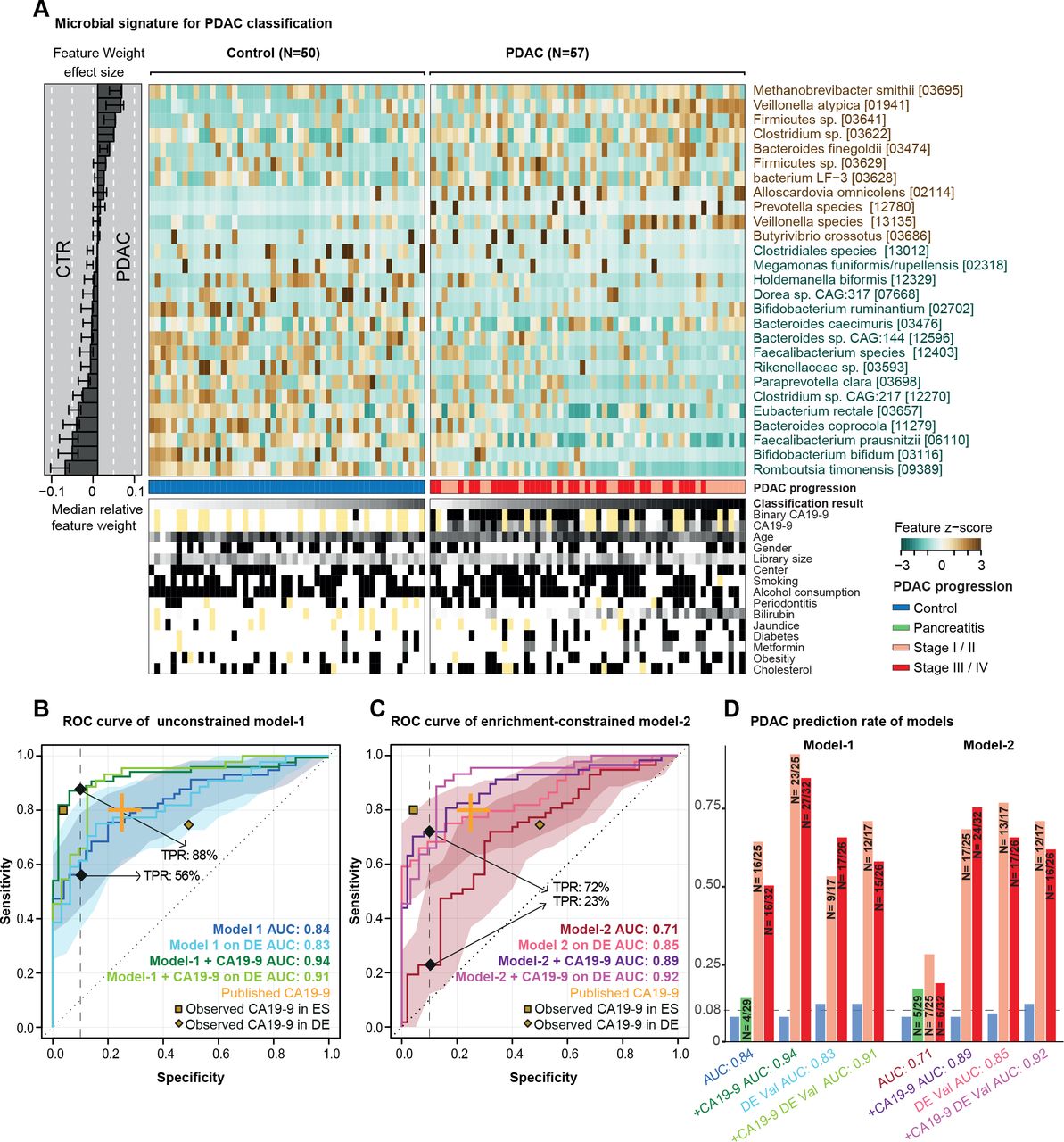
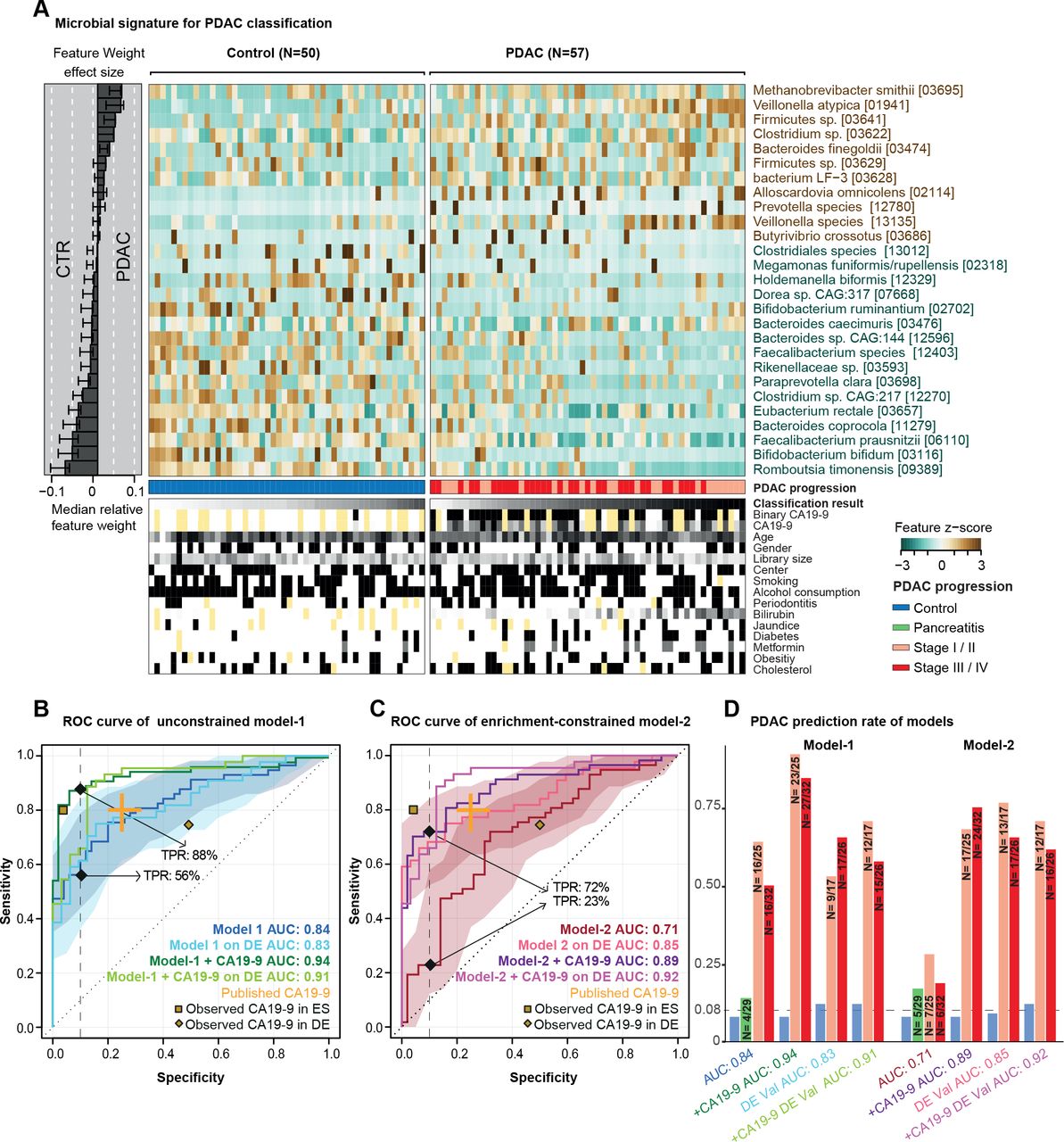
胰腺导管腺癌(PDAC)的预测微生物组特征。(A)样本粪便微生物群中27个选定物种的归一化丰度,显示为热图。右边的面板表示每个选定的特征对整体模型-1的贡献,每个特征的稳健性(将该特征作为预测因子纳入模型的百分比)以百分比表示。来自每个个体的交叉验证和被测试元变量的条件的分类分数显示在面板的底部,黄色表示缺失的信息。(B-D)无约束模型-1(无特征选择)、富集约束模型-2(被约束为阳性特征)和碳水化合物抗原(CA)19-9(阈值为37 μL/mL)与微生物特征的结合(见“方法”)的内部交叉验证结果显示为受试者工作特征(ROC)曲线,95% CI用相应颜色阴影表示。真阳性率(TPRs)以90%特异性临界值的百分比表示。所有模型在一个独立的德国(DE) PDAC测试总体(n=76)上的验证也被表示出来。发表的CA19-9准确性显示在橙色的元研究中。黄点代表我们人群中观察到的CA19-9准确性(西班牙(ES)人群中33/50对照组(CTRs)和44/57 PDAC患者的数据,德国(DE)人群中8/32 CTRs和44/44 PDAC患者的数据)(D)所有模型在不同PDAC进展阶段的TPRs,此外,慢性胰腺炎患者和对照组在90%特异性截点处的假阳性率显示为柱状图。由于总体样本量较低,所以将第一和第二阶段以及第三和第四阶段合并。 The number of predicted cases compared with the total is also shown on the top of each bar. DE-Val, German validation population.
第二种方法是在交叉验证(即针对每个训练集)中过滤特征,首先计算单个特征AUROC,然后删除AUROC <0.5的特征,从而选择PDAC(“富集约束”模型)中富集的特征。
为了将基于微生物的机器学习模型的预测与CA19-9标记相结合,将编码的CA19-9标记(1表示阳性,0表示阴性或不可用)添加到重复交叉验证运行的平均预测中,从而得到or组合。或者,通过将预测结果与CA19-9标记相乘计算AND组合。使用pROC R软件包v1.15计算两种组合的ROC曲线和AUROC值。7895% CI用对应的颜色表示,并在每张ROC曲线的图例中指定。
然后,在应用数据归一化例程后,将训练好的用于PDAC的ES宏基因组分类器应用于DE数据集,该例程选择相同的特征集,并使用相同的归一化参数(例如,使用SIAMCAT中的冻结归一化功能进行标准化的特征的平均值),与来自ES胰腺癌数据集的归一化过程相同。对于该分析,预测的分界点设置为初始ES PDAC研究人群中对照组的假阳性率为10% (图2).
数据预处理(过滤和归一化)、模型训练、预测和模型评估的所有步骤都使用SIAMCAT R包v.1.5.0执行79(https://siamcat.embl.de/).
宏基因组分类器的外部验证
为了评估训练模型的疾病特异性,我们从其他肠道宏基因组数据集(在线补充表S6)浏览完整的名单,包括登记号码)。我们进行了文献检索,以确定在相关疾病的病例对照或队列研究中公开获得的粪便宏基因组数据集。从欧洲核苷酸档案下载原始测序数据,并按上述方法进行分类分析,共25项研究,覆盖9个疾病州的5792个样本。35-59
然后,在应用数据规范化例程(该例程选择相同的特征集并使用相同的规范化参数(例如,通过使用SIAMCAT中的冻结规范化功能进行标准化的特征的平均值)后,将训练过的用于PDAC的宏基因组分类器应用于每个外部数据集。然后,对疾病特异性的预测进行评估,因为来自其他疾病样本的高预测分数表明分类器依赖于生物失调的一般特征,而不是胰腺癌特异性的信号,这不会导致来自其他疾病样本的假阳性率升高。对于该分析,预测的分界点被设置为初始PDAC研究人群中对照组的10%的假阳性率(图3).利用car R软件包v3.0-3中的cort .test函数(Spearman方法)检验25个居群的年龄、性别和测序深度对预测得分的影响。
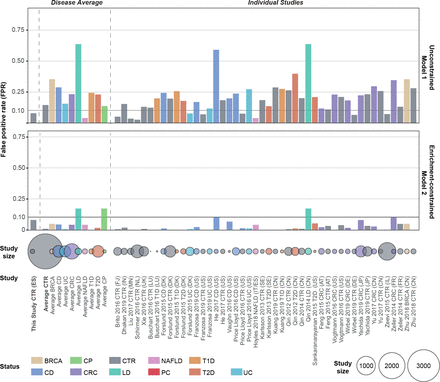
胰腺导管腺癌(PDAC)粪便微生物组模型疾病特异性的外部验证。25个外部测试集中宏基因组无约束模型-1和富集约束模型-2的假阳性率(FPR)显示为条形图(见在线补充表S4查看所有研究的列表)。验证数据集以与初始数据集相同的方式进行分析和规范化(参见“方法”)。根据健康状况对每项研究进行分层,并对模型进行测试,以在给定组中以90%的特异性截止值进行预测。来自其他疾病患者和健康个体的宏基因组的低FPR表明该模型是针对PDAC的。每一组的受试者数量以下面用颜色标注的圆圈显示。BRCA,乳腺癌;CRC,结直肠癌;克罗恩病;CP,慢性胰腺炎;CTR,对照组;LD,肝脏疾病; NAFLD, non-alcoholic fatty liver disease; PC, pancreatic cancer; T1D, type 1 diabetes; T2D, type 2 diabetes; UC, ulcerative colitis; ES, Spanish; DE, German.
亚种和菌株水平分析
宏基因组读数与proGenomes v1数据库中的物种代表基因组进行了映射80(见上图)。微生物单核苷酸变异体是由metaSNV的唯一定位读取而来,81样品之间的种内等位基因距离如前所述计算。82等位基因距离和PDAC疾病状态之间的关系在对潜在混杂因素(包括采样的身体部位)分层后使用PERMANOVA进行量化。
如前所述,定量菌株的口服-肠道传播。83简而言之,将受试者唾液和粪便样本中微生物单核苷酸变异的重叠与受试者之间的背景进行对比,以计算定量的口腔-粪便传播评分和p值。种类和主体特异性传播评分与临床因素的相关性采用方差分析和事后然后对多个测试进行Benjamini-Hochberg校正。
荧光原位杂交显微镜
FISH分析使用的探针专门针对特定细菌分类单元的16S rRNA序列(图4).所有的探针都是在文献搜索的基础上选择的,相应的分类显示在在线补充表S7).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
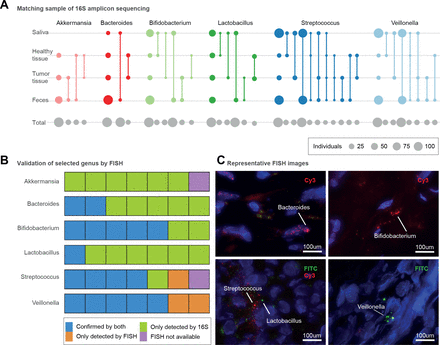
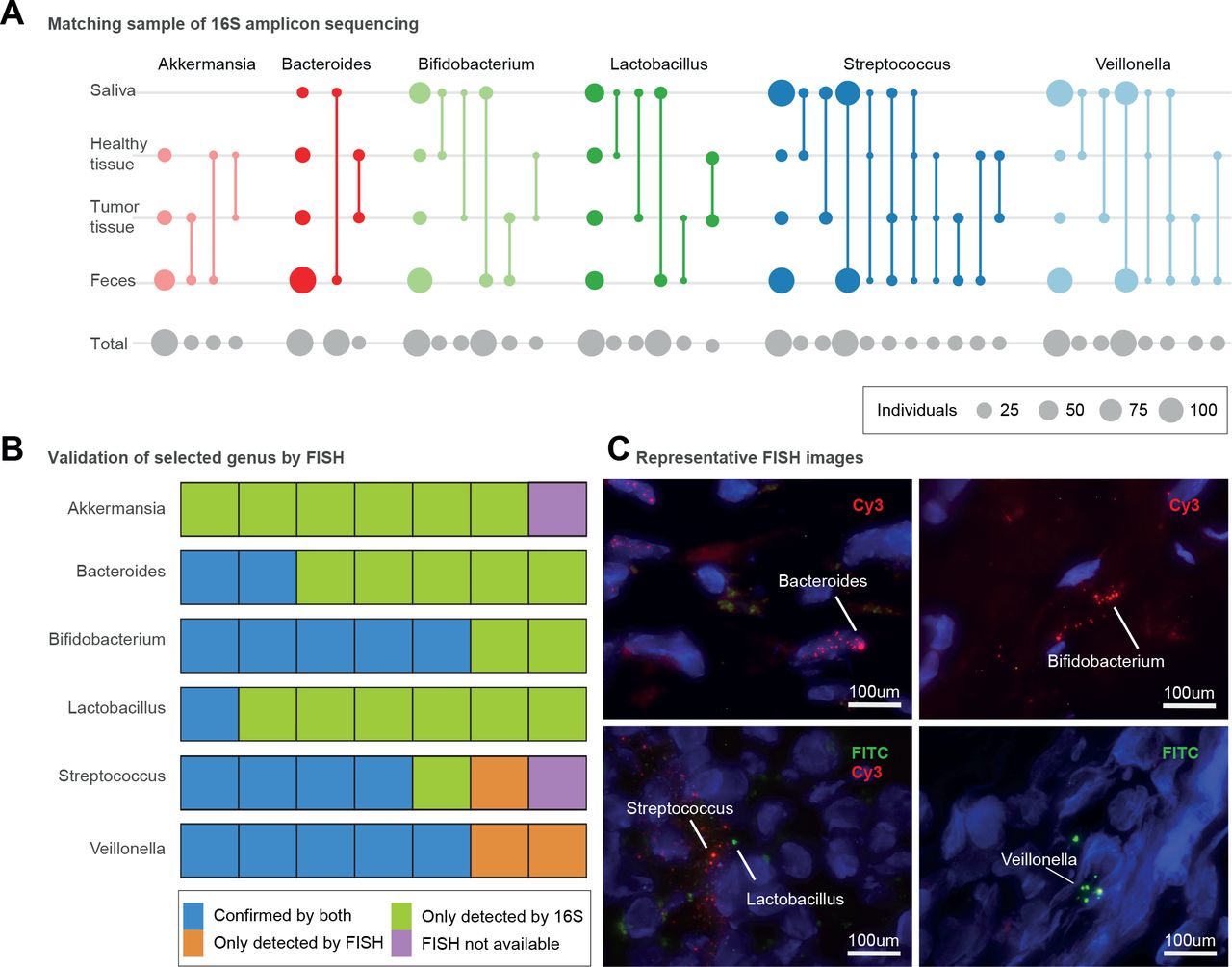
不同条件下胰腺不同部位的微生物群存在。(A)根据16S扩增子数据推断,在包括粪便、唾液、胰腺肿瘤和健康组织样本在内的四个不同身体部位存在不同属。圆圈大小对应于每次比较(灰色,底部行)或与内部个体匹配的扩增子序列变体(彩色)的受试者总数;匹配的样本类型用线连接。第一列显示在每个站点检测到该属的样本总数。(B) 7个选定的胰腺组织样本(5个肿瘤和2个非肿瘤)显示16S扩增子和荧光细菌存在/不存在原位杂交(FISH)方法。用16S扩增子测序和FISH验证细菌存在显示为蓝色。仅根据16S显示细菌存在的样品显示为绿色。仅通过FISH验证的细菌存在度显示为橙色,而由于缺乏组织材料而未进行FISH验证的样品显示为紫色。(C)具有代表性的显微镜图像拟杆菌(核内、肿瘤组织),双歧杆菌属(核外,肿瘤组织),乳酸菌(核外,非肿瘤组织),链球菌(核外,非肿瘤组织),韦永氏球菌属(核外,肿瘤组织)。采用异硫氰酸荧光素(FITC)和Cy3荧光染料,DAPI(4',6,-二氨基-2-苯基吲哚;蓝色)被用来标记细胞核。
从病理科取出胰腺肿瘤和正常胰腺样本,在手术切除后不到30分钟内立即在液氮中冷冻。无菌材料被用来解剖不同的样本。冷冻组织的最小尺寸约为0.125 cm3.(0.5×0.5×0.5厘米)。样品从临时液氮运输容器中转移,保存在-80°C的锁定冰箱中。在分析之前,将它们放在干冰上运输,移到液氮中最佳切割温度的模具中,立即在冷冻机上切割,得到10个3-5 μ m的切片。所有材料在每次样品处理后用乙醇消毒。
5 μ m厚度的组织切片安装在带正电的载玻片上(SuperFrost, Thermo Scientific)。简单地说,组织被固定在新鲜制备的4%多聚甲醛中。通过溶菌酶处理(10 g/L Tris HCl 6.5M)增强菌壁渗透性后,样品在杂交机(DAKO)中,在45°C下,在特异性探针存在下杂交1小时。在20 μ L的杂交缓冲液(20 nM Tris, pH 8.0)中进行杂交。在100ng的探针中加入0.9 M NaCl, 0.02%十二烷基硫酸钠,30%甲酰胺)。最后,组织在洗涤液(70%甲酰胺,10 mM Tris pH7.2和01%牛血清白蛋白)中洗涤,在一系列乙醇样品中脱水,风干,用0.5µg/mL DAPI(4’,6,-二氨基-2-苯基林多)/抗褪色溶液(Palex Medical)染色。捕获FISH图像使用徕卡DM5500B显微镜,CCD相机(Photometrics SenSys)连接到运行CytoVision软件7.2图像分析系统(应用成像)的PC机。对图像进行盲法分析,并根据探头信号的强度进行评分。
结果
当控制鸟枪宏基因组数据中的混杂因素时,PDAC与微生物组组成的适度变化相关
我们研究了57例新诊断,treatment-naïve例PDAC患者,29例慢性胰腺炎(CP)患者,以及50例年龄、性别和医院相匹配的对照组。参与者是在2016年至2018年期间从西班牙巴塞罗那和马德里的两家医院前瞻性招募的,使用相同的标准(见《受试者特征》)图1一个而且在线补充表S1-S3每个受试者的临床资料)。我们获得了所有受试者的粪便鸟枪宏基因组和45名PDAC患者、12名CP患者和43名对照组的唾液宏基因组(见“方法”)。分析工作流程详见在线补充图1.
因为一些PDAC的危险因素,如吸烟、饮酒、肥胖或糖尿病,本身与微生物组的组成有关84,我们首先试图在我们的研究人群中建立微生物组特征的潜在混杂因素,以便相应地调整分析。对于26个人口统计学和临床变量,我们量化了微生物组社区水平多样性的边际效应(在线补充表S4).当考虑到最常见的PDAC风险因素并应用0.05的错误发现率阈值时,粪便和唾液微生物组丰富度(作为alpha多样性的代理)与任何测试变量或PDAC状态无单变量关联(在线补充图2,在线补充表S4).
相比之下,微生物群落组成随诊断时的年龄(样本间bry - curtis差异的PERMANOVA, R2=0.01, benjamini - hochberg校正p=0.03)、粪便中的糖尿病(R2=0.01, p=0.04)和黄疸状态(R2=0.02, p=0.009)以及唾液中使用阿司匹林/扑炎息痛(R2=0.02, p=0.04)而变化,尽管效应量非常低(在线补充表S5).即使病例和对照组在年龄和性别上是匹配的,我们也将这些因素作为后续分析的层次。在这样的调整下,受试者的疾病状态是轻微的,但在统计学上显著与粪便中的群落组成相关(R2=0.02, p=0.001),但与唾液中的群落组成无关(R2=0.01, p=0.5) (图1 b,在线补充图3-4,在线补充表S5).事实上,PDAC患者的粪便微生物组组成与对照组(R2=0.02, p≤0.0001)和CP患者(R2=0.02, p=0.003)不同,尽管同样存在非常小的效应量。
高精度宏基因组分类器捕获PDAC患者的特定粪便微生物组特征
在总体群落组成的粗水平上建立了PDAC的肠道微生物组信号后,我们下一步确定了9个具有疾病特异性单变量关联的物种(PDAC病例与对照组相对丰度的Wilcoxon检验,benjamini - hochberg校正p<0.05;看到图1 c).最显著,韦永氏球菌属atypica,梭菌属nucleatum / hwasookii而且Alloscardovia omnicolens在PDAC患者的粪便中富集,而timonensis, prausnitzii粪杆菌,coprocola拟杆菌而且双歧杆菌bifidum物种集群被耗尽。相比之下,我们没有检测到唾液微生物组中有明显丰度差异的任何物种,在校正多个测试时,包括先前报道的关联,如牙龈卟啉单胞菌,放线菌聚集杆菌,22奈瑟氏菌属elongata或链球菌18(在线补充图5).
在单变量相关的粪便物种中,有几种自身对PDAC状态具有中度预测作用(在线补充图5).为了将这些个体信号合并到一个整体模型中,我们接下来通过10倍交叉验证拟合LASSO逻辑回归模型构建了多物种宏基因组分类器(见“方法”)。当不应用进一步的约束条件时,得到的模型在我们的研究人群中具有较高的准确性(“模型-1”;AUROC = 0.84;图2).模型中最显著的阳性标记物种为Methanobrevibacter smithii,Alloscardovia omnicolens,韦永氏球菌属atypica而且拟杆菌finegoldii.我们注意到,通过设计,LASSO回归在相互相关的集合中选择具有代表性的特征;因此,这些物种可能是具有高度相关丰度的较大物种集的代表。描述我们研究人群的26个人口统计学和流行病学变量中没有一个被模型选择为预测特征,而微生物组特征比任何其他特征都更有信息性(见在线补充图6和7).此外,这些变量中没有一个单独与模型中所代表的微生物物种相关,排除了它们作为潜在混杂因素的可能性。这表明分类器捕获了PDAC的诊断肠道微生物组特征,该特征可能独立于其他疾病风险因素和潜在混杂因素。
为区分CP患者和对照组而建立的类似模型没有预测能力(AUROC=0.5;在线补充图8),这与观察到的这些群体在成分上基本上难以区分一致。同样,唾液微生物组没有检测到强有力的PDAC特征(AUROC=0.48;在线补充图9).然而,区分PDAC患者和CP患者的粪便模型表现更好,其AUC为0.75,但CP组的样本量低限制了模型的鲁棒性(在线补充图8).我们进一步探索了功能微生物组剖面的高分辨率预测关联。基于KEGG模块丰度的模型(在线补充图10)达到了高达0.74的AUROC精度,但特征选择在验证折叠中同样不健壮,这是针对有限的样本集拟合大量变量(模块)的结果。因此,我们采用基于物种的分类器,因为它们提供了稳定的模型。
最初的基于肠道微生物的分类器包括与对照相比PDAC中缺失的几种物种,如普氏粪杆菌,拟杆菌,两歧双歧杆菌或Romboutsia timonensis(图2 b).对于其中的一些物种,以前的研究认为,通常情况下,蛋白质消耗与肠道炎症有关,而不是与特定的疾病有关。85因此,我们对分类器进行了重新训练,约束条件是在每个交叉验证褶皱中专门选择正相关(富集的)微生物特征。得到的富集受限模型(模型-2)识别PDAC患者的AUROC准确性为0.71。与无约束模型(模型-1)的差异主要归因于敏感性上的惩罚——即,在稀疏数据训练时,PDAC患者的自信检出率下降,与预期一致。
宏基因组分类器与抗原CA19-9水平的结合提高了准确性
血清中抗原CA19-9的水平被常规用于监测PDAC的进展,86 87但也被认为是PDAC早期诊断的潜在标记物,尽管报道的敏感性(0.80,95% CI 0.72 ~ 0.86)和特异性(0.75,95% CI 0.68 ~ 0.80)中等。12我们的西班牙人群中有77个个体(33/50对照组和44/57 PDAC患者)的CA19-9血清水平(在线补充图S11).鉴于CA19-9直接由肿瘤分泌,我们假设由CA19-9血清水平和我们的微生物组分类器提供的读数是互补的,它们的结合可以提高PDAC预测的准确性。事实上,考虑到CA19-9,我们的无约束模型-1的准确性从AUROC=0.84提高到0.94,主要是由于灵敏度的提高(图2 b).更引人注目的是,当我们用CA19-9信息修改富集约束模型-2时,我们观察到准确性从AUC=0.71大幅提高到0.89,同样是由于灵敏度的显著提高,因此基本上消除了相对于模型-1的性能损失(图2 c,在线补充图S11).在ES或DE人群中,在疾病晚期均无显著偏向于较高的CA19-9水平(在线补充图S11).
我们的西班牙研究人群包括25例早期(T1, T2) PDAC患者和32例晚期(T3, T4)受试者。疾病阶段不影响基于微生物的模型的性能(图2 d);特别是,回忆并不倾向于后期阶段。
基于宏基因组的分类器的性能推广到独立的验证队列
为了测试观察到的微生物组特征是否可以推广到西班牙研究人群之外,我们接下来在两个验证场景中挑战了我们的模型。首先,我们从德国埃尔兰根和法兰克福两家医院招募了44名PDAC患者和32名匹配的对照者作为独立研究人群,测试了预测的准确性图1、方法及在线补充表S3),这些样本的处理方法与西班牙人口的相同。在此DE验证总体上,无约束模型-1 (图2 b)和富集约束模型-2 (图2 c)的准确性与训练人群相当,甚至更高,无论是否有CA19-9水平的补充,且不同疾病阶段的趋势相似(图2 d).
接下来,为了确认我们的宏基因组分类器捕获的是pdac特异性特征,而不是非特异性的、更普遍的疾病相关变异,我们进一步针对各种健康状况的独立外部宏基因组数据集对它们进行了验证。总的来说,我们对来自18个国家25项研究的5792个公开可用的肠道宏基因组进行了分类,包括患有CP(本研究)、1型或2型糖尿病、结直肠癌、乳腺癌、肝病、非酒精性脂肪性肝病(包括克罗恩病和溃疡性结肠炎)的受试者,以及健康对照组(图3而且在线补充表S6).
当调整到聚焦ES研究人群的90%特异性(允许10%的假阳性预测)时,无约束模型-1显示ES人群中56%的PDAC患者和DE验证人群中的48%(假阳性率为6%),当补充CA19-9水平信息时(可用于8/32对照组和43/44 DE队列中病例的患者),可达64%。然而,模型-1的疾病特异性是有限的,在所有外部数据集中,平均预测15%的对照受试者的PDAC状态。大多数假阳性呼叫是在两个中国克罗恩病患者群体中观察到的48或者肝硬化。44克罗恩病与我们的模型中观察到的耗竭特征相似(特别是f . prausnitzii88),而肝病与胰腺功能受损有一些相同的生理特征。然而,所有其他肝病和克罗恩病组的误检率较低,表明这种影响可能部分归因于研究之间的技术和人口统计效应。事实上,我们注意到这两个中国研究人群的受试者明显比我们的人群年轻(Qin_2014的受试者为50±11岁;He_2017为28.5±8年;ES人群70±12耳)。这种年龄效应是系统性的:在所有验证集中,PDAC预测得分与受试者年龄相关(方差分析p=0.007;ρ斯皮尔曼= 0.16),以及与受试者的性别(p<106;)和测序深度(p=0.0008;ρ斯皮尔曼= 0.1) (在线补充图S12,在线补充表S6).
富集受限模型-2在两组PDAC患者中检出率较低,尽管CA19-9联合模型恢复了召回。模型2对PDAC具有高度特异性,在几乎所有外部人群中平均只有0-5%的PDAC预测,在上述人群中预测的最大值为17%44有肝病的人群。特别是,检测到的微生物组特征对2型糖尿病患者的错误分类也很有效(<2%的假阳性率);这与潜在的筛查应用有关,因为这些患者是PDAC的主要风险组(图3).
PDAC含有特有的细菌,与口腔和肠道微生物群落一致
胰腺分泌的改变,作为胰腺管中肿瘤生长的结果,可以影响消化功能,因此可能是肠道微生物组特征的基础,如上述所述。这意味着PDAC进展可以间接导致微生物组转移(即反向因果关系)。此外,胰管直接与十二指肠相通,为细菌提供了解剖链接25 30 89和真菌34在胰腺上定居并导致癌变。31
因此,我们假设在胰腺肿瘤中可以检测到与PDAC相关的几种肠道微生物类群。我们使用16S rRNA扩增子测序对研究人群中PDAC患者的所有粪便和唾液样本,以及肿瘤(n=23)和邻近健康胰腺组织(n=20)的活检进行了分类分析,并应用严格的过滤器排除了在细菌生物量低的样本中常见的假定试剂污染物33 90(见“方法”)。我们观察到胰腺微生物组惊人地丰富和多样化,在≥25%的样本中至少有13个细菌属,主要包括粪便微生物组中具有PDAC特征的类群91(图4一个,在线补充图13).在这其中,乳酸菌spp,Akkermansia muciniphila而且拟杆菌相对于非肿瘤胰腺组织,肿瘤中spp含量较高(Wilcoxon检验,错误发现率校正p<0.006)。
在5个肿瘤和2个非肿瘤胰腺组织样本的子集中,我们可以进一步验证患病率Akkermansiaspp,乳酸菌spp,双歧杆菌属仕达屋优先计划、韦永氏球菌属仕达屋优先计划,拟杆菌spp和链球菌spp使用带有属特异性引物的FISH分析(在线补充图4,在线补充表S7).一般来说,扩增子和FISH数据是一致的,但基于扩增子的检测似乎更敏感,可能是由于所分析的组织的数量。有趣的是,然而,Akkermansia尽管在26/30名受试者中通过扩增子测序观察到spp,但在所有测试样本中使用FISH均未检测到spp (图4 c,在线补充图14).
口腔、肠道和胰腺微生物群之间的联系
接下来,我们追踪了受试者唾液、粪便、肿瘤和健康组织样本中的扩增子序列变异(ASVs)。图4一),以使用16S rRNA数据可获得的最高分类学分辨率。韦永氏球菌属spp在PDAC患者的粪便中富集,在整个研究人群的唾液(100%的受试者)和粪便(87.5%)样本中高度流行,而口腔和粪便类型也匹配肿瘤和非肿瘤组织asv。有趣的是,我们没有发现个体内部匹配韦永氏球菌属肿瘤和邻近组织样本之间的ASVs,表明肿瘤存在韦永氏球菌属SPP可能与健康组织中的SPP不同。此外,我们的数据证实了先前的报告乳酸菌仕达屋优先计划26而且双歧杆菌属仕达屋优先计划25存在于PDAC肿瘤和非肿瘤组织中。对于这两个属,我们发现肿瘤类型对应于口腔或粪便ASVs,但不是两者都对应,而没有来自健康组织的ASVs与粪便样本匹配,这表明不同的胰腺亚群可能与口腔和肠道有关。
使用配对的唾液和粪便鸟枪宏基因组,我们进一步证实粪便pdac相关微生物菌株可能来自口腔(在线补充结果).
讨论
早期发现PDAC仍然是一个巨大的挑战,是减轻这种癌症负担的持续努力的核心。目前,fda唯一批准的PDAC生物标志物是血清CA19-9,主要用于疾病监测而不是筛查,由于敏感性和特异性的固有限制:CA19-9水平可能在一些与胰腺癌无关的情况下升高,而缺乏Lewis-A抗原的受试者根本不会产生CA19-9。10 - 12小规模研究提出了基于胰腺组织的PDAC标记,5尿液6 7和血清8 9适用性有限。然而,目前在临床中还没有PDAC的筛查工具,特别是在疾病早期阶段。
在一项前瞻性招募的新诊断研究人群中,对treatment-naïve名患者和匹配的对照组进行了口腔、粪便和组织微生物群分析(图1一个),我们开发了宏基因组分类器,仅基于特征粪便微生物物种,可以稳健而准确地预测PDAC (图2).我们的多物种模型捕获的PDAC特征与已确定的PDAC风险因素正交(图1B及2A).这表明,在实践中,粪便微生物组可用于筛选PDAC,作为其他可测试标记的补充,在联合试验中具有更高的诊断准确性,就像对结直肠癌提出的那样。39事实上,我们的微生物组分类器与CA19-9数据的组合(可用于我们人群的一个子集)显著提高了PDAC检测的准确性(图2罪犯).
先前的研究已经探索了PDAC和口腔之间的联系18-22 26 92 93或粪便23日24在16S rRNA测序的有限分类学分辨率下,对微生物组的关联模式提供了相互矛盾的报告,可能是由于不同的实验和分析方法。一些PDAC数据集的原始序列和患者水平的临床数据的不可用性使研究之间的比较具有挑战性,因此,PDAC相关微生物组特征的共识迄今尚未出现。一些先前报道的单变量口腔类群PDAC的相关性包括牙龈菌,放线菌,美国酸奶而且梭菌属SPP在我们的研究人群中未得到证实(在线补充图4);我们通常没有观察到任何唾液PDAC的特征,无论是单个物种或多物种模型。
我们仔细检查了人口统计学、生活方式和临床混杂因素的分析,因为这些可以显示出比疾病状态更强的微生物组相关性。84我们还验证了我们的宏基因组分类器针对独立采样,但一致处理的DE人群(图2罪犯)以及来自25个不同研究的不同健康状况的外部人群(n=5792)35-59(图3).当评估预测模型的疾病特异性时,混杂因素控制和外部验证都是必要的,特别是像PDAC这样在一般人群中发病率较低的疾病。这在我们的分析中得到了证实:在我们的两个宏基因组分类器中,模型-1在我们的ES研究人群中显示了AUROC=0.84的高准确性,这是由PDAC患者的高回忆率驱动的。然而,模型-1在外部验证中只显示了有限的疾病特异性,捕获了病例和对照组之间的非特异性物种耗尽信号,但也被患有其他疾病的受试者共享。这些包括一般的炎症特征,例如F. prausnitzii, E. rectale或b . bifidum.发表的各种疾病的宏基因组分类器,特别是先前报道的PDAC标记,都有类似的局限性:对焦点人群的高度调谐精度,但与其他疾病共享的非特异性特征。这种特异性的缺乏限制了它们在临床应用中的转化。相比之下,我们的模型-2由于受PDAC丰富特征的限制,在我们的人群中仅获得中等精度(ES上的AUC=0.71, DE上的AUC=0.85),但在外部人群中具有很高的PDAC特异性,错误预测率很低,包括已知的PDAC风险组,如2型糖尿病患者。特别是,在模型1和模型2中,PDAC富集特征与其他癌症类型(如结直肠癌)的粪便微生物组特征几乎没有重叠,这表明我们的微生物组模型与CA19-9水平(高度敏感,但不特异于PDAC)的结合是有希望的。我们注意到外部种群的残留假阳性率可能部分是由于技术异质性,因为所有外部种群的采样和处理都使用独立的协议,而且单个物种的单变量PDAC关联可能是有信息的,但不是针对疾病的(补充讨论)。因此,模型2中PDAC富集的物种显示了基于微生物的PDAC筛选的潜力,考虑到结合血清CA19-9的补充信息显著提高了准确性(AUC=0.89和0.92)。
我们的模型在PDAC疾病的各个阶段显示了相当的表现,对晚期没有偏见(图2罪犯).这表明在疾病发展过程中早期出现了特征性的微生物组特征,粪便微生物组可用于PDAC的早期检测。
我们的数据是严格观察和横断面的。尽管如此,有强烈的迹象表明,所确定的粪便微生物群变化不仅仅是胰腺功能受损或其全身影响的结果,尽管不能排除间接影响。在肠道和胰腺之间可以找到几个分类群,肿瘤相对于邻近健康组织的单变量富集,表明PDAC与肠道微生物组直接相关。我们证实了之前的观察25 30 31 89 91通过扩增子测序和FISH对迄今为止最全面的类群面板(图4).胰腺组织和肿瘤只含有少量细菌,因此容易在16S rRNA扩增子数据中受到污染33然而FISH检测需要特定的假设,因此对健康和患病胰腺微生物组组成的全面分类仍在出现。在我们的研究中,我们仔细过滤了我们的数据集,以对抗已知的套件污染物,并使用FISH测试确认了各种关键属的存在。此外,我们还观察到口腔、粪便和组织样本之间扩增子序列变体的个体内部重叠,证实了在扩增子数据可达到的最高分类学分辨率下,几个物种在多个地点共享存在。
因此,具有pdac相关分类群特征的粪便种群可以追溯到胰腺肿瘤。同样,我们观察到PDAC患者口腔-肠道菌株传播水平显著增加,特别是PDAC特征类群,表明这些可能来自个体内部,从口腔(在线补充结果).这些发现表明,口腔、肠道和胰腺微生物群可能是错综复杂的联系,像这里提出的多体位点研究设计是必要的,以理清它们各自在PDAC病因学中的作用和相互作用。
总之,所述粪便微生物组特征为PDAC检测提供了强大的宏基因组分类器,具有较高的疾病特异性,是现有标记的补充,并具有低成本的PDAC筛查和监测的潜力。此外,鉴于之前关于小鼠模型和人类中微生物介导的胰腺癌发生的报道,25 30 94我们相信,所展示的pdac相关细菌种类可能超越了它们的诊断用途,为疾病预防和治疗干预提供了有前途的未来切入点。
数据可用性声明
数据可在一个公共的、开放访问的存储库中获得。所有与研究相关的数据都包含在文章中或作为补充信息上传。样本的原始测序数据可在欧洲核苷酸档案(ENA)中获得,研究标识号为PRJEB38625和PRJEB42013。这些示例的元数据见补充表S1和S2。作为统计建模管道输入的过滤的分类和功能概要可在补充数据S1和S2中获得。分析代码和结果可在https://github.com/psecekartal/PDAC.git.
伦理语句
病人同意发表
伦理批准
参与者从马德里Ramón y Cajal医院和西班牙巴塞罗那Vall dHebron医院前瞻性招募。机构审查委员会伦理批准(CEI PI 26 2015-v7)和书面知情同意分别来自参与中心和研究参与者。在埃尔兰根大学医院外科(32个PDAC和32个对照样本)和歌德大学法兰克福诊所内科I部转化肝脏科(12个PDAC样本)招募了一个独立的验证人群。该研究获得了当地伦理委员会的批准(SGI-3-2019, 451_18 B)。临床数据,包括疾病分期和随访数据,从各自患者的医院图表的临床记录中检索。
致谢
我们感谢博克、马拉特和泽勒小组成员的鼓舞人心的讨论和所有贡献。此外,我们感谢EMBL基因组学核心设施对测序的支持。
参考文献
脚注
NM和PB是联合资深作者。
推特@ps_ecekartal, @TSBSchm, @o__maistrenko, @ZellerGroup, @JonelTrebicka, @nmalats, @BorkLab
EK、TSBS和EM-M贡献相当。
合作者PanGenEU研究调查员。西班牙国家癌症研究中心(CNIO),西班牙马德里:Núria Malats, Francisco X Real, Evangelina López de Maturana, Paulina Gómez-Rubio, Esther Molina-Montes, Lola Alonso, Mirari Márquez, Roger Milne, Ana Alfaro, Tania Lobato, Lidia eststudillo。意大利维罗纳大学:Rita Lawlor, Aldo Scarpa, Stefania Beghelli。爱尔兰国家癌症登记处,科克,爱尔兰:Linda Sharp, Damian O’driscoll。马德里-北桑奇纳罗医院,马德里,西班牙:曼努埃尔·伊达尔戈,Jesús Rodríguez帕斯夸尔。西班牙马德里拉蒙·卡哈尔医院:阿尔弗雷多·卡拉托,亚历杭德拉·卡米诺亚,卡门Guillén-Ponce,梅赛德斯Rodríguez-Garrote,费德里科Longo-Muñoz,雷耶斯·费雷罗,凡妮莎Pachón, M Ángeles瓦兹。西班牙巴塞罗那德尔玛医院:Mar Iglesias, Lucas Ilzarbe, Cristina Álvarez-Urturi, Xavier Bessa, Felipe Bory, Lucía Márquez, Ignasi Poves, Fernando Burdío, Luis Grande, Javier Gimeno。西班牙巴塞罗那Vall dHebron医院:Xavier Molero, Luisa Guarner, Joaquin Balcells, Mayte Salcedo。德国慕尼黑工业大学:Christoph Michalski, Irene Esposito, Jörg Kleeff, Bo Kong。瑞典斯德哥尔摩卡罗林斯卡学院:Matthias Löhr,黄佳琪,Caroline Verbeke,叶伟民,余静茹。 Hospital 12 de Octubre, Madrid, Spain: José Perea, Pablo Peláez. Hospital de la Santa Creu i Sant Pau, Barcelona, Spain: Antoni Farré, Josefina Mora, Marta Martín, Vicenç Artigas, Carlos Guarner, Francesc J Sancho, Mar Concepción, Teresa Ramón y Cajal. The Royal Liverpool University Hospital, UK: William Greenhalf, Eithne Costello. Queen’s University Belfast, UK: Michael O’Rorke, Liam Murray, Marie Cantwell. Laboratorio de Genética Molecular, Hospital General Universitario de Elche, Spain: Víctor M Barberá, Javier Gallego. Instituto Universitario de Oncología del Principado de Asturias, Oviedo, Spain: Adonina Tardón, Luis Barneo. Hospital Clínico Universitario de Santiago de Compostela, Spain: Enrique Domínguez Muñoz, Antonio Lozano, Maria Luaces. Hospital Clínico Universitario de Salamanca, Spain: Luís Muñoz-Bellvís, J.M. Sayagués Manzano, M.L. Gutíerrrez Troncoso, A. Orfao de Matos. University of Marburg, Department of Gastroenterology, Phillips University of Marburg, Germany: Thomas Gress, Malte Buchholz, Albrecht Neesse. Queen Mary University of London, UK: Tatjana Crnogorac-Jurcevic, Hemant M Kocher, Satyajit Bhattacharya, Ajit T Abraham, Darren Ennis, Thomas Dowe, Tomasz Radon. Scientific advisors of the PanGenEU Study: Debra T Silverman (NCI, USA) and Douglas Easton (U. of Cambridge, UK).
MAGIC(专注于微生物的德国跨学科合作)研究研究者。法兰克福:Jonel Trebicka, Hans-Peter Erasmus, Fabian Finkelmeier, Robert Schierwagen, Wenyi Gu, Olaf Tyc, Frank Erhard Uschner, Stefan ZeuzemGreifswald大学外科:Stephan Kersting, Melanie Langheinrich。Erlangen大学外科:Robert Grützmann, Georg F. Weber, Christian Pilarsky。埃尔兰根大学内科:Stefan Wirtz。
贡献者EK设计研究,进行实验工作,获取和分析数据,撰写初稿和修改稿。TSBS设计研究,获取和分析数据,撰写初稿和修改稿。新兴市场-M designed the study, contributed to patient recruitment and the collection of biomaterials and clinical data, acquired and analysed data, and wrote the first manuscript draft.SR-P contributed to patient recruitment and the collection of biomaterials and clinical data and conducted experimental work.JW, OMM, WAA, BAA, AC, HP-E, FF, PG-R, SKe, ML, MM, XM, RT-R, JT contributed to patient recruitment and the collection of biomaterials and clinical data. RJA, AF, AMG, KZ contributed to data analysis. LE contributed to patient recruitment and the collection of biomaterials and clinical data and conducted experimental work. RH, FJ, SKa, AT conducted experimental work and acquired data. AO, TvR contributed to data analysis. MSI, PSI contributed to patient recruitment. VB acquired data. GZ designed the study and contributed to data analysis. FXR designed the study and contributed to data analysis and wrote the first manuscript draft. NM conceived the study, designed the study, contributed to patient recruitment and the collection of biomaterials and clinical data and wrote the first manuscript draft. PB conceived of the study, designed the study, contributed to data analysis and wrote the first manuscript draft. All authors reviewed, edited and approved the final version of the manuscript.
资金我们感谢EMBL、CNIO、世界癌症研究中心(# 15-0391)、欧洲研究理事会(ERC-AdG-669830 MicrobioS)、bmbf资助的德国生物信息学基础设施网络(de. nbi #031A537B)内的海德堡中心人类生物信息学中心(HD-HuB)、西班牙卡洛斯III-FEDER Salud Carlos III-FEDER卫生调查基金会(FIS)的资助(资助号PI15/01573、PI18/01347、FIS PI17/02303);红色Temática de Investigación Cooperativa en Cáncer,西班牙(批准号为RD12/0036/0034, RD12/0036/0050, RD12/0036/0073);III beca Carmen Delgado/Miguel Pérez-Mateo de aesppan - acanpan;EU-6FP集成项目(#018771-MOLDIAG-PACA);EU-FP7-HEALTH (# 259737 - canceralia)。资助者没有参与研究设计、患者登记、分析、手稿撰写或审查。
相互竞争的利益EK、TSBS、JW、OMM、EM-M、GZ、LE、SR-P、FXR、NM和PB已申请基于微生物生物标志物的胰腺癌早期检测专利(申请号:EP21382876.7)。其他作者声明没有利益冲突。
来源和同行评审不是委托;外部同行评审。
补充材料本内容由作者提供。它没有经过BMJ出版集团有限公司(BMJ)的审查,也可能没有经过同行评审。讨论的任何意见或建议仅仅是那些作者(s)和不被BMJ认可。BMJ放弃从放在内容上的任何依赖产生的所有责任和责任。如果内容包含任何翻译材料,BMJ不保证翻译的准确性和可靠性(包括但不限于当地法规、临床指南、术语、药品名称和药物剂量),并且不对翻译和改编或其他原因引起的任何错误和/或遗漏负责。
请求的权限
如果您希望重用这篇文章的任何部分或全部,请使用下面的链接,它将带您访问版权清除中心的RightsLink服务。您将能够快速获得价格和以多种不同方式重用内容的即时许可。