








文摘
检测体点替换描述癌症基因组是一个关键的一步。然而,现有的方法通常小姐low-allelic-fraction突变发生在只有一个子集的测序细胞由于肿瘤异质性或由正常细胞污染。我们现在MuTect,方法,贝叶斯分类器适用于检测分数非常低的体细胞突变等位基因,只需要一些支持读取,紧随其后的是仔细调整过滤器,确保高特异性。我们还描述基准测试方法,使用真实的,而不是模拟,测序数据评价敏感性和特异性的函数测序深度、基础质量和等位基因的一部分。与其他方法相比,MuTect具有更高的敏感性与特异性,尤其与等位基因突变分数低至0.1及以下,使得研究癌症MuTect特别有用积累及其进化在标准外显子组和基因组测序数据。
主要
体单核苷酸替换是一个重要和常见的癌症中改变基因功能的机制。然而,他们是很难确定的。首先,他们发生在基因组中一个非常低的频率,从0.1到100突变megabase (Mb),根据肿瘤类型1,2,3,4,5,6,7。第二,改变只可能出现在的一小部分来自特定基因位点的DNA分子的原因包括污染正常细胞样本,分析当地癌症基因组和人类基因组变异的突变的肿瘤细胞只在群8,9,10,11(“subclonality”)。DNA分子窝藏一个变更的一部分(“等位分数”)已经低至0.05高度不洁净的肿瘤8。肿瘤的subclonal结构的研究不仅是理解肿瘤演化的关键在疾病进展和对治疗的反应12也为开发可靠的个性化的癌症治疗的临床诊断工具13。
最近的报告subclonal事件在癌症已经使用三个不同的非标准实验策略:(i)分析克隆突变出现在几个,但不是全部,同一个病人的转移,这表明这些突变subclonal原发肿瘤14;(2)超深测序检测subclonal突变11;或(3)测序很小数量的单个细胞15,16,17。相比之下,成千上万的肿瘤被测序标准深度100 - 150为外显和30 - 60××整个基因组作为大型癌症基因组项目的一部分,比如癌症基因组图谱1,2,7国际癌症基因组协会18。检测克隆和subclonal突变出现在这些示例,需要一个高度敏感和特定mutation-calling方法。虽然特异性可以控制通过后续实验验证,这是一个昂贵和耗时的步骤,是不切实际的通用应用程序。
任何体细胞mutation-calling方法的敏感性和特异性沿着基因组不同,取决于几个因素,包括肿瘤的深度序列覆盖率和patient-matched正常样本,当地测序错误率,等位基因的突变和证据阈值用于声明一个突变。描述敏感性和特异性取决于这些因素对于设计实验是必要的和足够的权力来检测突变在给定的等位基因的分数,以及推断沿着基因组突变的频率,这是理解的关键参数突变分析过程和意义19,20.。
为了满足这些高敏感性和特异性的关键需求,不足够重视可用方法21,22,23,我们开发了一个调用者的体细胞突变点,MuTect。在其发展,MuTect被用在许多协作研究1,2,3,4,7,19,24,25,26,27,28,29日,30.,31日,32,33,34,35。这里我们描述MuTect的公开版本,包括其背后的基本原理不同的组件。我们也估计其性能作为上述因素的函数使用基准测试方法,据我们所知,没有描述。性能的方法是在先前的研究也支持独立的实验验证3,4,7,19,24,25,26,27,28,29日,30.以及它的应用程序在其他出版物的数据集分析36。我们证明我们的方法是几次比其他方法更敏感low-allelic-fraction事件同时保持非常具体,允许深入探索突变的高度不洁净的肿瘤样本和景观subclonal肿瘤的进化。
MuTect是免费为非商业使用http://www.broadinstitute.org/cancer/cga/mutect(补充数据)。
结果
基准评估变异呼叫者
许多mutation-detection方法已经开发出来,但很少有系统的方法对真正的测序数据基准测试他们的表现。以前的出版物描述了模拟方法从完全合成的模型21那些更好的捕捉真正的测序错误11。然而,这些方法模型完整的多样性的非随机测序错误的参考和备选等位基因在基因组的网站。为了更好地评估mutation-detection性能的方法,我们使用两种基准测试的方法,将采样和“虚拟肿瘤”。
Downsampling使用主要测序数据的读取验证子集体细胞突变来测量的敏感性的突变调用者标识已知的突变。子集生成由实验得到的数据集随机扣除读取直到到达所需的深度报道。值得注意的是,将采样保持预期的原突变等位基因的分数,因为读取被不管是否包含突变的等位基因。将采样的方法是有限的在四个方面:(i)验证事件的数量通常是小,灵敏度估计导致较大的误差;(2)因为等位基因保留分数,只有之前验证等位基因分数可以探索;(3)分析排除了任何没有最初发现的突变,因此可能高估了真正的敏感性;及(iv)特异性无法衡量。
将采样来解决这个问题,我们开发了一个基准测试程序,包括创建“虚拟肿瘤”,我们都知道真正的突变与确定性(在线方法和补充图1)。测量特异性,我们创建了虚拟肿瘤和正常的数据集,在控制深度,从测序数据中生成两个不同的测序实验相同的正常样本(指定样本)。所有的突变确定必然是假阳性。测量灵敏度,我们模拟的体细胞突变等位基因控制分数通过替换选择读取virtual-tumor数据集与读取第二个样本(指定样品B)在位点是引用和示例B港口高信任度细胞杂合的事件。然后,我们评估的能力一个算法来检测这些模拟的体细胞突变。通过这种方式,我们可以测量灵敏度使用真实的测序数据所需的深度覆盖和等位基因的一部分。
两种基准测试的方法是互补的。将采样使用真正的体细胞突变,但有限的参数政权它可以用来探索,它不能直接用来测量特异性。相比之下,virtual-tumor方法没有这些限制。然而,它使用带有事件模拟体细胞突变,不同于体细胞突变的核苷酸替换的频率和上下文。调整基地品质变化为不同的基地(由于偏差在机器错误),有变量的检测灵敏度不同的替换(补充图2)。因为灵敏度的差异很小,我们选择使用所有的细胞活动。然而,随着virtual-tumor方法可以模拟特定的肿瘤类型的突变谱带有事件重新匹配预期的突变谱的肿瘤。
检测与MuTect体细胞突变
MuTect采取作为输入序列数据匹配的肿瘤和正常读取参考基因组的DNA后对齐和标准预处理步骤38,39,40,其中包括标记的重复读取,校准基质量分数和局部调整。每个独立基因位点的方法运作,由四个关键步骤(图1):(i)的低质量的序列数据(补充的方法);(2)变异检测肿瘤样本中使用贝叶斯分类器;(3)过滤去除假阳性产生的相关序列不是被错误的工件模型;及(iv)指定的变异体细胞或带有一个贝叶斯分类器。

变异检测
变异的肿瘤识别的数据分析数据在每个站点上在两个替代模型:(i)参考模型,米0假设没有变异的网站和任何观察nonreference基地是由于随机测序错误,和(2)一个变体模型, ,假设站点包含真正的等位基因变体米在等位基因的一部分f除了测序错误。等位基因分数f是未知的,估计分数tumor-sample读取的支持吗米。这种显式建模的f,而不是假设杂合的,二倍体事件,使我们的方法比其他方法更敏感21,22。我们声明米候选人变体的数据如果对数似然比变体和参考模型(即日志赔率(LOD)得分)超过一个预定义的阈值决定,取决于预期的突变频率和期望的假阳性率(在线方法)。的选择决定阈值可以用来控制特异性和敏感性之间的权衡,描述由接受者操作特征(ROC)曲线(图2一个)。我们使用固定阈值6.3的结果提出除非另有指示。这个阈值对应于一个106.3:1优势比的参考模型,这是合理的,因为在许多肿瘤突变的频率只有1 - 10 / Mb,因此网站的先验概率窝藏突变可能低至1:105或1:106。
,假设站点包含真正的等位基因变体米在等位基因的一部分f除了测序错误。等位基因分数f是未知的,估计分数tumor-sample读取的支持吗米。这种显式建模的f,而不是假设杂合的,二倍体事件,使我们的方法比其他方法更敏感21,22。我们声明米候选人变体的数据如果对数似然比变体和参考模型(即日志赔率(LOD)得分)超过一个预定义的阈值决定,取决于预期的突变频率和期望的假阳性率(在线方法)。的选择决定阈值可以用来控制特异性和敏感性之间的权衡,描述由接受者操作特征(ROC)曲线(图2一个)。我们使用固定阈值6.3的结果提出除非另有指示。这个阈值对应于一个106.3:1优势比的参考模型,这是合理的,因为在许多肿瘤突变的频率只有1 - 10 / Mb,因此网站的先验概率窝藏突变可能低至1:105或1:106。

(一个)的敏感性和特异性MuTect突变的等位基因分数0.2,肿瘤样本测序深度30×30×和正常样本测序深度的使用各种的LOD值阈值(θT)(≤0.1θT≤100)。计算敏感性和假阳性率使用独立的测序错误的模型和均匀Q35基质量分数和准确的阅读位置(计算)以及结果显示virtual-tumor方法标准(MuTect STD)和高信任度(MuTect HC)配置。一个典型的设置θT= 6.3使用黑色圆圈标记。(b)敏感性肿瘤样本测序深度的函数和等位基因分数(f)使用θT= 6.3。计算灵敏度的一个以及结果显示virtual-tumor方法和验证将采样的结直肠突变7。误差线,95%置信区间(通常小于标志)。
LOD分数是有用的作为一个阈值检测,观察到的一致性virtual-tumor预测灵敏度和测量灵敏度的方法(图2)。尽管如此,LOD分数不能立即译成一个变种的概率是由于真正的突变而不是测序错误因为LOD评分计算在一个假设的独立测序错误和准确的阅读位置。我们在下面讨论,这些假设不正确,因此,虽然直接应用的LOD分数准确估计敏感性检测突变,它大大低估了假阳性。
变体过滤
消除这些额外的假阳性由于不准确的阅读位置和诱致性排序错误,我们开发了六个过滤器(图1和表1)。此外,我们使用正常样本的面板控制,消除细胞活动和工件(在线方法)。这些过滤器的子集定义多个版本的方法(图1):(i)标准(STD),适用于没有过滤器,因此包括所有检测到变异;(2)高信任度(HC),适用于六个过滤器和(iii)高信任度+面板的正常样本(HC +彩球),此外应用“正常样本面板”(其)过滤器。
我们测试了这些过滤器通过应用他们的效用virtual-tumors基准和recomparing结果计算(图2一个)。的灵敏度估计(HC)和没有(STD)过滤器相似,表明该模型是准确检测和过滤不不利影响的敏感性。然而,在应用过滤器(HC)、特异性增加,密切关注计算,表明过滤器在很大程度上消除系统的误报(图2一个和补充图3)。
变体的分类
最后,每个变体中发现肿瘤样本被指定为体细胞(不是出现在匹配的正常样本),细胞(出现在匹配的正常样本)或变体(肿瘤样本中但不确定的状态与正常样本不足的结果数据)。执行这个分类中,我们使用一个LOD分数比较的可能性的数据在模型变体存在杂合子或缺席在匹配的正常样本(在线方法)。我们声明,如果有足够的数据分类能力进行细胞分类还不到95%。我们也使用公共细胞变异数据库41先验概率的事件被细胞。
灵敏度
我们应用几种基准测试方法来评估我们的方法的敏感性检测突变等位基因测序深度和分数的函数(图2 b)。首先,我们计算模型下的灵敏度独立测序错误和准确的阅读位置使用我们的统计检验给定一个等位基因的部分和肿瘤样本测序深度,并假设所有基地有固定Q35基质量分数(近似意味着基质量分数在模拟数据;网上的方法和补充图4)。
接下来,应用downsampling基准,我们使用3753年验证体细胞突变,分层的等位基因分数(值= 0.28,= 0.07 - -0.94),在结肠直肠癌7deep-coverage (≥100×), exome-capture测序数据从数据库下载的基因型和表型(dbGAP;phs000178)。最后,应用virtual-tumor基准测试中,我们使用deep-coverage数据从两个高覆盖率,全基因组样本(柯瑞尔个人NA12878和NA12981)测序Illumina公司HiSeq仪器作为1000人基因工程的一部分42和另一个先前的研究43,1 Gb的基因组区域。注意,我们不能用彩球过滤器(HC +彩球)virtual-tumor敏感性指标,因为它丢弃常见细胞网站。
灵敏度估计基于这三种方法相互高度一致(每个深度平均变异系数为3.1%)。这表明基准测试方法准确地估计mutation-calling方法的灵敏度也计算灵敏度是健壮的在一个大范围的参数值,使我们能够自信地推断分数较高的测序深度和较低的等位基因(补充表1)。
基于这种分析,我们发现MuTect是一个高度敏感的检测方法。它检测到突变在肿瘤的网站30×深度数据(典型的全基因组测序)和一个等位基因的一部分0.2灵敏度为95.6%。深度测序的敏感性增加到99.9%(50×)降至58.9%,检测突变等位基因的分数为0.1(30×测序;图2 b和补充表1)。有150×测序深度(典型的外显子组测序)我们观察到66.4%的敏感性为3%等位基因部分事件。正是这种敏感性检测low-allele-fraction事件独特位置MuTect分析样品纯度较低的或复杂的subclonal结构。
这详细了解确定敏感性因素对于目标的适当深度测序是至关重要的。因为一个突变的等位基因的分数取决于肿瘤的纯洁,本地拷贝数和单克隆8,一个可以计算所需的测序深度所需的敏感性肿瘤特异性的基础上。同时,给定一组测序数据我们可以计算灵敏度的检测与特定等位基因突变分数为每个基地以及基因组。这让我们坚持没有突变的情况下(有特定的等位基因的一部分),这是特别重要的在临床设置。
特异性
是简单的创建一个极度敏感的体细胞突变信号检测方法通过识别任何网站用一个突变nonreference解读为候选人。显然,这种方法会导致一个巨大的假阳性率。因此在评估mutation-detection方法的性能,关键是彻底描述其特异性。假阳性的有两个来源:(i)为肿瘤overcalling事件数据和(2)undercalling真正带有匹配的正常事件数据。Overcalling肿瘤数据通常是由于测序错误和不准确的阅读位置,而真正的带有undercalling事件与正常样本通常是由于低测序深度在正常样本。
测量肿瘤的假阳性率由于overcalling数据,我们使用了virtual-tumor方法在1 Gb NA12878序列数据在不同深度在虚拟肿瘤和正常30×在虚拟样本。所有检测到的事件是假阳性,但从考虑消除那些undercalling细胞所产生的事件,我们排除了所有已知的细胞变体网站。使用没有过滤器(STD),假阳性率随深度增加(从6.7 Mb−1在5×覆盖率为20.1 Mb−130×覆盖;图3)。这是由于增加的力量叫突变等位基因分数较低,富含假阳性(图3 b)。HC过滤器误判率降低了一个数量级(1.00 Mb−1在30×报道)。彩球过滤器(HC +彩球)然后过滤掉大部分剩余的罕见,但复发,工件(0.51 Mb−1在30×报道)。某些过滤器,如“可怜的映射”过滤器,在低深度影响最大,而其他过滤器更不变的测序深度的变化,如“近端差距”过滤器(图3 c)。集群的位置的过滤拒绝大多数网站只。然而,大部分的假阳性拒绝了几个过滤器。

(一个)体细胞误称错误率为真正的参考站点作为肿瘤样本测序深度的函数性病,HC和HC +彩球MuTect配置。虚线,所需的假阳性。误差线、95%的置信区间。(b)等位基因的分布比例为所有误称作为肿瘤样本测序深度的函数。(c)的事件被每个过滤器;散列区域表示拒绝事件完全是由每个过滤器。(d)体细胞误称错误率真正带有杂合的单核苷酸多态性网站正常样本的测序深度时,网站是变体的人口(dbSNP)和未知的(不是dbSNP)。误差线、95%的置信区间。(e,f)权力测序深度的函数在正常样本分类这些事件在未知的细胞或体细胞(e),(f)细胞变体网站。
然后我们研究了真正的错误由于undercalling带有事件匹配的正常样本,而是使用相同的方法∼100万年相同的领土(细胞变异位点图3 d-f)。将一个事件划分为细胞或体细胞,MuTect常见细胞变异位点的使用不同的先验概率与其余的基因组,因此我们报告假阳性率分别为这两个场景(图3 d)随着权力对此类事件进行了分类(图3 e, f)。我们观察到≤7读取以前未知的细胞在正常数据变化网站(图3 e)或≤18读取已知细胞变异的网站(图3 f),没有足够的数据分类的一个变种是体细胞或细胞,因此我们一直等网站“变体”,从来没有假阳性体细胞在这些情况下。一旦有足够的数据分类,错误率下降迅速从2.4×10−3在8×覆盖在正常样本低于0.2×10−3在12×覆盖率,对应不到一个更进一步的胚系事件在整个外显子组(∼30 Mb的外显子组×50前所未知的细胞变异Mb−1×0.2×10−3出错率)。
最后,我们使用MuTect在最近的一些研究发现一致的验证率∼95%基于多个正交编码区域验证技术3,4,7,19,24,25,26,27,28,29日,30.(表2)。这些研究使用了MuTect的早期版本中,较不敏感,但在一个出版13使用本文描述的版本的MuTect等位基因突变出现在7%分数102阅读(8)被发现,随后由超深测序验证(∼6000×报道)。事实上,验证速度并不是最好的衡量误判率进行比较研究,因为它取决于假阳性比真正的突变,这因肿瘤类型而异。我们也因此报告假阳性率本身(表2)。我们观察到假阳性中值的0.16 Mb−1低于我们报道,使用全基因组数据(图3),但符合测量速率限制分析时编码区域(补充图5),这表明编码区域更容易测序和比对错误。
相比其他方法
我们使用了将采样和virtual-tumor基准测试方法比较与其他常用方法:MuTect SomaticSniper21,JointSNVMix22和Strelka23。我们测试每个方法在两个配置,标准(STD)和高信心(HC),与阈值选择方法产生相似的误判率。SomaticSniper (v1.0.0),我们使用了发布配置。JointSNVMix (v0.7.5),我们使用的检测阈值P(体细胞)≥0.95的性病PHC(体细胞)≥0.9998。0.4.7 Strelka(版本),我们使用的推荐配置质量分数≥15 HC和质量分数≥1性病。
我们评估方法的敏感性对等位基因分数和肿瘤样本测序深度使用virtual-tumor (图4),将采样(补充图6灵敏度)方法,观察到一个明显的区别,特别是在较低的等位基因分数。我们分析数据为30×序列覆盖率。在标准配置,所有方法显示≥99.3%敏感性的突变等位基因分数为0.4。然而,在HC配置、MuTect JointSNVMix和Strelka仍然敏感(分别为98.8%、96.6%和98.5%),而SomaticSniper敏感性下降到91.5%。0.1一个等位基因的一部分,MuTect HC发现超过一半的变异(53.2%),而Strelka HC, JointSNVMix HC和SomaticSniper HC检测到29.7%,分别为16.8%和7.4%的突变。在一个更低的0.05等位基因的一部分,MuTect HC敏感性16.0%但这60×覆盖率提高到51.9%。相比之下,JointSNVMix HC和SomaticSniper HC的敏感性≤2.0%,敏感性并没有增加与肿瘤样本测序深度明显。Strelka HC发现只有4.6%的30×事件报道,只有60×覆盖率提高到20.8%。灵敏度等low-allelic-fraction事件描述不洁净的关键在异构的肿瘤,肿瘤或subclonal突变,在这个政权MuTect更敏感。

(一个)敏感性肿瘤样本测序深度的函数和变异等位基因分数(f)表示mutation-detection方法和配置。(b)体细胞误称错误率为真正的胚系网站测序深度的函数在正常样本。(c为真正的参考站点)体细胞误称错误率肿瘤样本测序深度的函数。虚线,所需的假阳性。(d)敏感性的函数特异性突变的等位基因分数0.1,肿瘤样本测序深度30×30×和正常样本测序深度显示方法和配置。黑色虚线显示变化之间的敏感性和特异性性病和HC配置方法。灰色的实线是MuTect virtual-tumor方法的结果补充图3。误差线,95%置信区间(一个- - - - - -c)。
作为一个更敏感的方法也不太具体,我们也的表现方法相比对两种假阳性。我们观察到一个非常低的假阳性率由于miscalled胚系网站的所有方法给出足够的深度(≥15×)与正常样本(图4 b)。假阳性利率每megabase由于miscalled参考网站(图4摄氏度)可比高于20×覆盖在STD配置(值= 10.2,= 0.7 - -20.1)和HC配置(值= 1.0,= 0.2 - -3.1)。
我们可以总结为每个方法的敏感性和特异性之间的权衡使用ROC曲线,这取决于测序深度在肿瘤和正常样本和突变等位基因分数。在图4 d我们举个例子使用肿瘤样本测序深度30×,正常样本测序深度30×0.1等位基因的一部分,显示MuTect通常是一个更敏感的对于一个给定的特异性和降低灵敏度也有一个小得多的一个类似的增加特异性所获得的HC配置。
我们也比较了灵敏度的方法使用之前报道的测序数据和验证突变科罗拉多州- 829黑素瘤细胞系37(补充表2)。尽管MuTect略比其他方法更敏感,这个数据集代表纯粹的细胞系,很容易被high-allelic-fraction事件(值= 0.55),因此不暴露方法之间的区别。通过运行MuTect和其他突变打电话我们发现额外的突变不是最初报道(补充表3和4),强调比较突变在文献中报道通常低估了灵敏度为完整的地面实况组体细胞突变往往是未知的。
讨论
随着新体细胞突变呼叫者的发展,癌症基因组学社区将大大受益于使用这里描述的方法系统的测量性能在整个参数空间的肿瘤和正常样本在不同的测序深度和突变等位基因分数。我们的方法以及工具我们开发基准mutation-detection方法可用,我们鼓励开发人员报告他们的方法使用这些指标的特点。这里描述的方法可以扩展到其他病变如insertion-deletions (indels)或重组。
我们的数据表明,MuTect优于其他方法的特异性和敏感性(之间的权衡图4)。敏感的优势MuTect来源于variant-detection统计测试,其中包括等位基因的一个评估分数的事件和工作点选择沿着ROC曲线。SomaticSniper JointSNVMix使用一个模型基于克隆突变纯,二倍体肿瘤(因此假设一个固定的等位基因分数50%)。这种假设降低了低灵敏度等位基因部分事件。相比之下,Strelka特别考虑等位基因的一部分,因此在STD MuTect配置有相似的敏感性。然而,当在推荐的HC运行配置控制假阳性,MuTect只有轻微下降敏感性与其他方法相比。这可能是因为过滤器在MuTect仔细调整拒绝真正的假阳性调用不牺牲敏感性。
我们发现MuTect比竞争更敏感的在给定的特异性方法,使我们能够更全面地描述体细胞突变的风景,尤其是那些在肿瘤细胞的一小部分。此外,这可以用标准的测序深度,使大型数据集的分析,全球正在生成。分析subclonal癌细胞突变和分数的变化,港口是一种强大的方法来研究进展治疗期间积累的进化,转移和复发11,12,44,45。特别是,我们证明了存在subclonal突变基因在慢性淋巴细胞白血病是一个独立的预后因素超出了当前使用的临床参数13。使用标准的外显子组测序数据,我们发现突变出现在低至10%的癌症细胞,代表预计0.05等位基因的一部分(假设一个二倍体地区的杂合突变)之前占污染基质,并发现这些突变似乎有影响的时间第一次治疗13。
low-allelic-fraction因为其他方法不敏感事件,他们可能会因此错过重要subclonal司机进展或阻力。因此,MuTect的敏感性检测subclonal突变等位基因较低分数是一个实质性的进步,未来的关键发现关于癌症的subclonal架构和翻译这些发现的临床诊断影响癌症患者治疗和结果。
方法
Virtual-tumor基准测试方法。
virtual-tumor方法始于deep-coverage从高覆盖率数据,样本(NA12878)全基因组测序Illumina公司HiSeq工具:两个库42、“solexa - 18483”和“solexa - 18484”, 30×每和一个图书馆43“solexa - 23661”, 30×。这些数据是公开的;的细节可以在补充表5。
首先,我们随机测序数据划分为多个分区。我们创建了6个分区分别从三个库(18)分区,因此创建数据分区∼5×覆盖。我们可以通过排序BAM完成这一点39的名字使用SortSam皮卡德(http://picard.sourceforge.net/)工具来有效地读取随机排序。然后我们随机分配每个读到一个分区和分区特定BAM文件写的。
测量特性,我们可以指定某些分区一样“肿瘤”和其他“正常”,和过程通过MuTect(或其他方法)。体细胞突变确定在这个过程是假阳性细胞undercalled事件在正常或错误的变异导致测序噪音overcalled分区指定为肿瘤。我们画读从图书馆- 18483和- 23661 solexa solexa肿瘤样本和正常样本库solexa - 18484的。
测量灵敏度,我们求助于第二个人(额外的测序数据补充表5)。在这种情况下,我们选择了NA12891,也是测序60×1000人基因组计划的一部分。使用发表高信任度单核苷酸多态性(SNP)基因型的样本1000人基因工程,我们识别出一套杂合的的网站在NA12878 NA12891和纯合子的参考。然后我们使用另一个实用工具,SomaticSpike, MuTect软件包的一部分,执行一个混合的实验在网上。在每个选定的网站,这个实用程序试图取代的读取由二项分布使用指定的等位基因的分数从NA12891 NA12878数据读取数据,因此模拟体细胞突变的位置、类型和预期的等位基因的一部分。如果没有足够的读入NA12891替换所需的读入NA12878,网站被跳过。这个过程的输出是一个虚拟的肿瘤BAM与在网上变异和一组位置的变异。然后灵敏度估计试图在这些网站检测突变。
变异检测。
为每个网站我们表示参考等位基因r∈{一个,C,G,T}表示,b我和e我的称为基础阅读我(我= 1…d),涵盖了网站和误差的概率基本调用(每个基地都有一个关联的Phred-like质量分数问我在哪里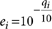 )。叫一个变种在肿瘤我们使用两个模型试图解释数据:(i)模型米0现场没有变异和nonreference基地都解释为测序噪音,和(2)模型
)。叫一个变种在肿瘤我们使用两个模型试图解释数据:(i)模型米0现场没有变异和nonreference基地都解释为测序噪音,和(2)模型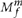 等位基因的变体米真正在现场存在等位基因的一部分f,如米0,阅读也受到测序噪音。请注意,米0相当于
等位基因的变体米真正在现场存在等位基因的一部分f,如米0,阅读也受到测序噪音。请注意,米0相当于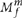 与f= 0。
与f= 0。
该模型的可能性 是由
是由

假设在读取测序错误是独立的。如果所有替换等可能的错误,即发生的概率e我/ 3,我们获得
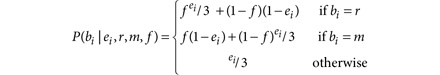
变异检测执行通过比较两种模型的可能性,如果他们的比例,也就是说,LOD分数,超过一个决策阈值(日志10δT我们声明米作为候选人变体。我们计算
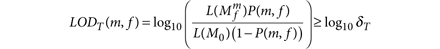
并设置δT2,以确保我们至少两倍相信这个网站是变体相比,噪音。我们也可以重写LODT作为

来确定P(m f),我们首先假设P(米),P(f)是统计独立的P (f)均匀分布(即P(f)= 1)P(米)预计三分之一的突变频率为研究肿瘤类型(代表平等之前替换)。在实践中,我们使用一个典型的3×10的突变频率−6的收益率θT= 6.3。
我们发现的最大LODT在所有的三个值米和未知的等位基因部分参数设置f我们可以使用最大似然估计,也就是说,找到f最大化LODT。然而,对于计算效率,我们估计 作为
作为
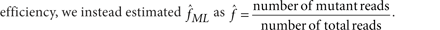
一种常见的假阳性肿瘤DNA的突变调用污染与其他个体的DNA。带有单核苷酸多态性的DNA污染表现为体细胞突变。我们先前已经表明,这种污染能产生许多假阳性和开发了一个工具,比赛46估计的污染水平,f续在测序数据。低污染的DNA是一种常见的现象,甚至2%的污染可以产生166个假阳性电话每megabase和10个假阳性调用megabase当排除已知的SNP网站46。来防止这种类型的假阳性,使受污染的样品分析,我们与变体模型取代了参考模型,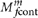 。这可以保证变异称为只有当他们也极不可能解释为污染。
。这可以保证变异称为只有当他们也极不可能解释为污染。
变体过滤器:面板的正常样本。
减少假阳性和miscalled细胞事件,我们使用一个面板的正常样本作为一个过滤器。创建这个过滤器,我们跑MuTect一组正常样本就像肿瘤样本没有匹配的正常样本在STD模式。从这些数据,为网站创建一个VCF文件被确认为变体MuTect在不止一个正常样本。
这VCF然后提供给调用者,拒绝这些网站。然而,如果网站中提供的VCF已知的已知突变它保留,因为这些网站可以代表复发体细胞突变,已发现在正常样本的面板正常样本来自相邻组织或有污染肿瘤DNA。
正常样本用于构造这个面板中,权力越高将检测和删除罕见的工件。因此,我们通常使用的所有正常的现成的样品。结果是通过使用一组全基因组测序数据从125年血液正常样本实体瘤患者癌症。样品使用的一部分virtual-tumor方法并不包括在这个面板。
变体的分类。
执行这个分类中,我们使用一个类似上面描述的一个分类器。在这种情况下,f在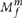 适当地设置为0.5细胞杂合的变体。因此我们有
适当地设置为0.5细胞杂合的变体。因此我们有

可以写成
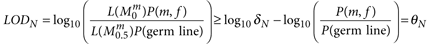
注意,这里的条件倒,因为我们想要相信改变是不存在。为δN10,我们设置一个阈值,超过阈值δT因为我们想要更有信心在我们的变体的分类分类错误的细胞事件将很快出现在下游体细胞分析由于其显著的提升人口频率周期性网站相比真正的体细胞事件。
计算P(生殖细胞系)我们区分两种情况:(i)网站已知变异在人口和(2)所有其他网站。我们使用公共dbSNP数据库41这种区别。
有∼30×106网站已知变异根据dbSNP释放134人口,这是∼1000个变异/ Mb。一个给定的个人通常∼3×106在他们的基因变异,其中95%在dbSNP网站41,42。因此我们希望∼50变异/ Mb不是dbSNP网站,也就是说,P(生殖细胞系| non-dbSNP网站)= 5×10−5因此我们使用θN | non-dbSNP网站= 2.2。在dbSNP网站,然而,我们预计的95%∼3×106变异发生在30×106网站dbSNP数据库,屈服P(生殖细胞系| dbSNP网站)= 0.095,因此θN | dbSNP网站= 5.5。
灵敏度计算。
计算灵敏度检测突变等位基因的一部分f使用n读取Phred-like质量分数问(因此一个基本错误,e的, ),我们首先计算k读取的最小数量的不同等位基因,将触发一个变体使用电话
),我们首先计算k读取的最小数量的不同等位基因,将触发一个变体使用电话

然后观察的概率灵敏度k或多个读取给定等位分数和深度。的边际分布读取与备用等位基因的数量,来自备用基地或误读的参考基础,遵循一个二项分布的频率反映了真正的底层等位基因的一部分f和错误的概率e(注意,这里我们把最坏的情况下,所有的误读基地转换为相同的替代等位基因)。因此我们可以计算的概率k或更多的读
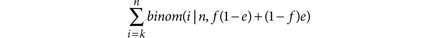
引用
癌症基因组图谱的研究网络。综合基因组分析卵巢癌。自然474年,609 - 615 (2011)。
癌症基因组图谱的研究网络。全面的基因组特征定义了人类胶质母细胞瘤基因和核心通路。自然455年,1061 - 1068 (2008)。
纳杰,s . et al .序列分析突变和易位的乳腺癌亚型。自然486年,405 - 409 (2012)。
Stransky: et al .头颈部鳞状细胞癌的突变景观。科学333年,1157 - 1160 (2011)。
叮,l . et al。体细胞突变影响肺腺癌的重要途径。自然455年,1069 - 1075 (2008)。
伯杰,年报等。黑色素瘤基因组测序揭示频繁PREX2突变。自然485年,502 - 506 (2012)。
癌症基因组图谱网络。全面的分子表征人类结肠癌和直肠癌。自然487年,330 - 337 (2012)。
卡特,S.L. et al .绝对量化体细胞的DNA改变在人类癌症。生物科技Nat。》。30.,413 - 421 (2012)。
沃尔特,M.J. et al .克隆继发性急性髓系白血病(aml)的体系结构。心血管病。j .地中海。366年,1090 - 1098 (2012)。
公园,林亭汝,Gönen, M., Kim, H.J., Michor, F. & Polyak, K. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype.j .中国。投资。120年636 (2010)。
Nik-Zainal, s . et al . 21乳腺癌的生活史。细胞149年,994 - 1007 (2012)。
叮,l . et al。克隆进化复发急性髓系白血病揭示了全基因组测序。自然481年,506 - 510 (2012)。
兰道,D.A. et al。进化,在慢性淋巴细胞白血病subclonal突变的影响。细胞提前在线出版,doi: 10.1016 / j.cell.2013.01.019(2013年2月14日)。
坎贝尔,P.J. et al .基因组不稳定性的模式和动态转移性胰腺癌。自然467年,1109 - 1113 (2010)。
纳文:et al .肿瘤进化推断单细胞测序。自然472年,90 - 94 (2011)。
侯,y . et al .单细胞外显子组测序和单克隆进化JAK2-negative骨髓增殖性肿瘤。细胞148年,873 - 885 (2012)。
徐,x et al .单细胞外显子组测序揭示肾肿瘤的单核苷酸突变特征。细胞148年,886 - 895 (2012)。
国际癌症基因组协会等。国际癌症基因组项目的网络。自然464年,993 - 998 (2010)。
查普曼,硕士et al。最初的基因组测序和分析多发性骨髓瘤。自然471年,467 - 472 (2011)。
斯坦利·g . et al .评论“共识”编码序列的人类乳腺癌和结肠直肠癌。科学317年1500 (2007)。
拉森,D.E. et al . SomaticSniper:体细胞点突变的识别在全基因组测序数据。生物信息学28,311 - 317 (2012)。
罗斯,a . et al . JointSNVMix:体细胞突变的概率模型进行准确探测正常/肿瘤配对下一代测序数据。生物信息学28,907 - 913 (2012)。
桑德斯,比较温度et al . Strelka:准确的体细胞small-variant打来的测序tumor-normal样本对。生物信息学28,1811 - 1817 (2012)。
巴比里,刚建成时et al .外显子组测序识别复发SPOP, FOXA1和MED12基因突变在前列腺癌。Nat,麝猫。44,685 - 689 (2012)。
鲈鱼,A.J. et al .基因组测序的结直肠腺癌标识一个周期性VTI1A-TCF7L2融合。Nat,麝猫。43,964 - 968 (2011)。
王、l . et al . SF3B1和其他小说在慢性淋巴细胞白血病癌症基因。心血管病。j .地中海。365年,2497 - 2506 (2011)。
普,T.J. et al。成神经管细胞瘤外显子组测序揭示subtype-specific体细胞突变。自然488年,106 - 110 (2012)。
伯杰,年报等。人类前列腺癌主要的基因组的复杂性。自然470年,214 - 220 (2011)。
Lohr, J.G. et al .发现和优先级的体细胞突变弥漫型大b细胞淋巴瘤(DLBCL) whole-exome测序。Proc。国家的。学会科学。美国109年,3879 - 3884 (2012)。
Imielinski, m . et al .映射和大规模并行测序肺腺癌的特点。细胞150年,1107 - 1120 (2012)。
王,p . et al .突变异柠檬酸脱氢酶1和2经常发生在肝内胆管癌和恶性胶质瘤分享甲基化目标。致癌基因推进,在线出版,doi: 10.1038 / onc.2012.315(2012年7月23日)。
Durinck, s . et al .颞解剖肿瘤发生的主要癌症。癌症。1,137 - 143 (2011)。
李,R.S. et al。一个非常简单的基因组构成高度恶性儿科杆状的癌症。j .中国。投资。122年,2983 - 2988 (2012)。
癌症基因组图谱的研究网络。等,全面的鳞状细胞肺癌基因组特征。自然489年,519 - 525 (2012)。
hodi、大肠等。景观的司机突变黑色素瘤。细胞150年,251 - 263 (2012)。
福布斯,S.A. et al。宇宙:采矿完成癌症基因组的体细胞突变在癌症的目录。核酸Res。39D945-D950 (2011)。
游乐园,既有et al。综合目录人类癌症基因组的体细胞突变。自然463年,191 - 196 (2010)。
李,h &杜宾,r .快速和准确的短阅读符合burrows - wheeler变换。生物信息学25,1754 - 1760 (2009)。
李,h . et al .序列比对/地图格式和SAMtools。生物信息学25,2078 - 2079 (2009)。
DePristo,硕士et al。变化的框架使用下一代DNA测序数据发现和基因分型。Nat,麝猫。43,491 - 498 (2011)。
雪莉,明沟等人dbSNP: NCBI数据库的遗传变异。核酸Res。29日,308 - 311 (2001)。
1000人基因工程财团。从人口规模的测序人类基因组变异的地图。自然467年,1061 - 1073 (2010)。
Gnerre, et al。高质量的草案从大规模并行序列数据哺乳动物基因组的组装。Proc。国家的。学会科学。美国108年,1513 - 1518 (2011)。
Shah……等。在小叶乳腺肿瘤的突变进化描述单核苷酸的分辨率。自然461年,809 - 813 (2009)。
Yachida, s . et al .远处转移发生在胰腺癌的基因进化。自然467年,1114 - 1117 (2010)。
Cibulskis k . et al .竞赛:估计人类样本在新一代测序数据的交叉污染。生物信息学27,2601 - 2602 (2011)。
确认
这项工作是由美国国家卫生研究院资助U54HG003067 U24CA143845。我们感谢基因组分析工具包(GATK)集团,为他们的有价值的反馈和我们的测试用户。
作者信息
作者和联系
贡献
D.J.构成的概念使用统计方法和过滤器来检测体细胞突变。和MuTect构思和kc设计和分析。kc分析执行的算法和实现。M.S.L.构思和最初发达的彩球过滤器。调查和和S.L.C.开发能力计算与MuTect subclonal事件检测。C.S.响亮的协助验证数据的生成和解释。D.J.,C。S. and M.M. critically reviewed the manuscript. K.C., G.G. and E.S.L. wrote the manuscript. G.G., M.M., S.G. and E.S.L. led the project.
相应的作者
道德声明
相互竞争的利益
kc,G.G.和米。S.L. are inventors on US provisional patent application no. 61/693,987 covering the method described in the paper.
权利和权限
关于这篇文章
引用这篇文章
Cibulskis, K。,Lawrence, M., Carter, S.et al。敏感体点突变的检测不洁净和异构癌症样本。生物科技Nat》31日,213 - 219 (2013)。https://doi.org/10.1038/nbt.2514
收到了:
接受:
发表:
发行日期:
DOI:https://doi.org/10.1038/nbt.2514
本文引用的
基因组的特性及其在骨oligometastatic NSCLC的潜在含义
BMC肺药(2023)
测试开发、优化和验证遗传病的WGS管道
BMC医学基因组学(2023)
从门店数据验证基因变异使用卷积神经网络
BMC生物信息学(2023)
satmut_utils:多路检测的模拟和变体叫包变体的效果
基因组生物学(2023)
收养neoantigen-reactive T细胞疗法:改进战略和当前的临床研究
生物标志物的研究(2023)
