
摘要
结直肠癌是一种常见于老年人的癌症,通常由称为腺瘤的良性息肉缓慢发展而来。肠道菌群被认为直接参与结直肠癌的发生。然而,与腺瘤或癌相关的肠道微生物的身份和功能能力还没有得到全面的调查。在这里,我们对晚期腺瘤和癌患者以及健康受试者的粪便进行了宏基因组范围的关联研究(MGWAS),揭示了各组中富集的微生物基因、菌株和功能。对潜在风险因素的分析表明,相对于水果和蔬菜,大量摄入红肉似乎与细菌的生长有关,这可能会导致更不利的肠道环境。这些结果表明,基于粪便微生物的策略可能有助于结直肠腺瘤或癌的早期诊断和治疗。
简介
结直肠癌(CRC)是全球三大最常诊断的癌症之一,也是癌症死亡的主要原因之一1,2.较发达国家的发病率较高,但在历史上风险较低的地区,如东亚、西班牙和东欧,发病率迅速上升,原因是所谓的西方生活方式1,2,3..在结直肠癌的发展过程中,基因的改变累积了许多年,通常包括肿瘤抑制基因腺瘤性大肠息肉病(APC),然后激活和灭活突变喀斯特,PIK3CA而且TP53(参考文献3.,4).大多数CRC病例是散发的,但在此之前有发育不良的腺瘤,可发展为恶性形式,称为腺瘤-癌序列3..
结直肠癌是研究最多的与肠道菌群有关的疾病之一。然而,因果关系通常是通过应用抗生素鸡尾酒来研究的,这些鸡尾酒可以根除肠道微生物群,而不知道确切的微生物菌株和基因在起作用5,6,7.梭菌属与正常结肠组织相比,在大肠癌中是否检测到8,9,在腺瘤中富集10.梭菌属nucleatum一种牙周病原体,已被证明可促进肠道肿瘤的髓细胞浸润Apc分钟/ +小鼠和相关的促炎基因的表达增加,如Ptgs2(cox - 2),Scyb1(IL8),白细胞介素6,肿瘤坏死因子(肿瘤坏死因子α),Mmp3在老鼠和人类身上11.然而,尚不清楚是否更多的细菌或古生菌可以作为结直肠癌的标记物,或有助于结直肠癌的病因学。此外,也许是人类健康最重要的环境因素,或我们的“另一个基因组”12,13在美国,肠道微生物组是否以及如何整合其他风险因素,如饮食、吸烟、肥胖,仍有待探索1,2,3.,14,15并产生结肠直肠癌发生的相干信号。
在这里,我们展示了156份大肠癌患者和健康对照组的宏基因组鸟枪测序粪便样本,鉴定了宏基因组连锁群(MLG)。16肿瘤的特征,并揭示各种风险因素的可能影响,特别是红肉与水果和蔬菜的消费对结肠直肠腺瘤-癌序列肠道微生物的改变。
结果
肠道微生物群的全球变化
为了研究结直肠腺瘤和直肠癌患者肠道微生物组的变化,我们对156份健康对照、晚期腺瘤或癌症患者的粪便样本进行了宏基因组鸟枪测序(补充数据1).高质量的测序结果为(平均每个样本5gb,补充数据2)集合起来新创,鉴定出的基因被编译成一个包含350万个基因的非冗余目录,平均每个样本中76.3%的reads被绘制出来。
我们首先调查了三组肠道菌群的丰富性和均匀性(图1).对55例健康对照组、42例晚期腺瘤患者和41例癌患者的起始队列进行稀疏分析,结果显示各组基因丰富度接近饱和,晚期腺瘤高于对照组,癌高于晚期腺瘤(图1一个).三组间基因丰富度和属丰富度均有显著差异(P= 0.005,P= 3.2e-7,分别为Kruskal-Wallis检验;图1 b, e),而α-多样性(图1 c、f),与之前对腺瘤和健康对照的16S核糖体RNA基因焦磷酸测序分析一致17.毒力因子数据库中毒力基因的数量18各组之间也有显著差异(P= 1.2e-5, Kruskal-Wallis检验;图1 d,补充数据3).因此,在这个队列中,基因或属的更丰富并不是健康肠道菌群的标志,但可能表明各种有害细菌或古菌在晚期结直肠腺瘤或癌患者中过度生长。

肠型,肠道菌群的另一种一般测量方法19,20.,将队列分为两组或三组(根据所使用的方法,图2 a, b,补充图1a),每一个都包含健康对照、腺瘤和癌患者。然而,更大比例的癌和腺瘤患者的肠型中含有高水平的拟杆菌所代表的肠型中发现了更多的健康样本瘤胃球菌属(图2 c, d,补充图1b,c).既不是原来的分区围绕中间体(PAM)聚类方法19也没有基于Dirichlet多项混合模型的方法20.检测到一个普氏菌-显性肠型,符合人群特有特征或肠型的连续性21.分析证实,在结直肠癌发生之前或发生期间,肠道菌群发生了深刻的变化。

腺瘤或癌的特征
为了探索健康或肿瘤样本中肠道微生物组的特征,我们确定了130715个基因,这些基因在三组中的任何两组中表现出显著的丰度差异(Kruskal-Wallis测试,Benjamin-Hochberg测试)问值< 0.1;图3).除肿瘤状态外,在对照组、腺瘤患者和癌症患者中,除血清铁蛋白和红肉摄入量外,其他可用表型均无显著差异(P<0.05, Kruskal-Wallis检验,补充数据4).与健康和晚期腺瘤样本相比,癌中约58.9%的基因标记物显著升高(图3),表明它们是结直肠癌特有的;另有24.3%的基因在癌组织中明显高于对照组,在晚期腺瘤中处于中等水平。在有下降趋势的基因中,癌中5388个(占总数的4.1%)明显低于健康和晚期腺瘤样本;2601个(占总数的2.0%)在癌中明显低于对照组,在晚期腺瘤中处于中等水平。这些对照富集基因比腺瘤或癌富集基因更常被定位到京都基因和基因组百科全书(KEGG)通路。图3 b).增加和减少基因数量的差异表明,在癌的发展过程中,病原菌的增加比有益菌的减少更为明显。

(一个所有130,715个差异富集基因(Benjamin-Hochberg问-value<0.1)按照对照与腺瘤、对照与癌、腺瘤与癌的富集方向进行分组,其中+表示后者富集,−表示前者富集,0表示无差异(p≥0.05,Wilcoxon秩和检验,Bonferroni校正为对照进行多重检验)。各组基因的相对丰度用小提琴图表示,即四分位范围(IQRs;粗条形图)、中位数(条形图上的开放点)、第一和第三四分位1.5倍IQR内的最低和最高值(条形图上和条形图下的线)以及值的密度(曲线之间的宽度)。每组基因的数量显示在括号中,在中间的条形图上用彩色块表示。(b基因标记在KEGG level 2通路中的分布一个.
根据不同样本间丰度的共变异,将差异显著的基因聚类为mlg,从而确定各组的微生物物种特征16.一些拟杆菌而且Parabacteroides物种,还有Alistipes putredinis,Bilophila wadsworthia,Lachnospiraceae细菌而且大肠杆菌与健康和晚期腺瘤样本相比,在癌样本中富集(图4,补充数据5).可能的口服厌氧菌mlg-75, mlg-83, mlg-84, mlg-88(与梭菌属口腔分类单元370,Pavimonas微米,Gemella morbillorum而且消化链球菌属stomatis,分别)和mlg-77形成了一个正相关簇,相对独立于其他富癌mlg (图4 b, c).与对照组相比,腺瘤组织中Mlg-75、mg -88和mg -77水平也有所升高(图4).肠道共生体如Bifidobactium animalis而且乳酸链球菌另一方面,在腺瘤或癌症患者的粪便中,与健康微生物组的偏离一致。

(一个)对照与晚期腺瘤(n=55和42)。(b)晚期腺瘤与癌(n=42和41)。(c)对照与癌对照(n=55和41)。对于所有含有>100基因的mlg,富集方向采用Wilcoxon秩和检验(P< 0.05,补充数据3).MLG中基因数量(102~3613)随节点大小而变化。标记为种或属的mlg按科着色。节点之间的边表示根据比较样本计算的Spearman相关性>0.8(绿色),0.6到0.8(浅到深蓝色)或<−0.6(红色)。
与mlg一致,属包括瘤胃球菌属,双歧杆菌属而且链球菌的比例明显高于对照组,而拟杆菌,Alistipes,埃希氏杆菌属,Parvimonas,Bilophila而且梭菌属在癌症患者(补充图2).
大肠杆菌、mg -331、mg -711和mg -1607在组织学诊断为癌的样本中含量较高原位与腺癌的样本相比,mlg-75、mlg-83、mlg-84和mlg-1697在腺癌中的含量更高(P<0.05, Wilcoxon秩和检验,错误发现率(FDR)=0.7216;补充图3).126个mlg中有7个(包含超过100个基因)在癌分期中表现出显著差异22(P<0.05, Kruskal-Wallis检验,FDR=0.531),通常在II或III期达到峰值(补充图4).在直肠癌或左结肠(脾屈、降结肠和乙状结肠)癌患者的样本中,许多与癌相关的mlg比右结肠(盲肠、升结肠和横结肠)的样本更丰富(补充图5),这表明粪便是胃肠道末端环境的最佳代表,但仍可能显示结肠起始处的恶性肿瘤。
基于mlg的腺瘤或癌的分类
为了说明粪便微生物组对结直肠癌的诊断价值,我们构建了一个可以检测癌症样本的随机森林分类器。在由55个对照和41个癌症样本组成的训练集中,5次重复10次交叉验证(即50次测试)导致15个MLG标记物的最佳选择,这些标记物在训练集中表现良好(图5 a - c,补充数据1、5).在测试集(8个对照组,47个晚期腺瘤和5个癌)中,分类错误仍然很低,显示接受者工作曲线下的面积(AUC)为96%(晚期腺瘤被认为是非癌,图5 d, e,补充数据1).包括年龄和身体质量指数(BMI)以及126 mlg没有改变所选择的标记。大多数mlg在老年和中年受试者(65岁以上和65岁以下;补充图6,补充数据5),表明与癌症相关的肠道微生物群的共同特征。

(一个)随着mlg数量的增加,癌症随机森林分类中5个10倍交叉验证误差的试验分布。该模型使用对照和癌样本中MLGs(>100基因)的相对丰度进行训练。n=55和41)。黑色曲线表示五次试验的平均值(灰色线)。粉红色的线标记了最佳集合中mlg的数量(补充数据5).如果将年龄和BMI与mlg一起包括在内,则选择相同的mlg。(b中模型所示的交叉验证训练集中癌的概率的盒须图一个.(c)训练集的受试者工作曲线(ROC)。受试者工作曲线下面积(AUC)为98.34%,95%可信区间(CI)为96.29 ~ 100%。(d)测试集的分类包括8个对照(绿色),47个晚期腺瘤(蓝色)和5个癌(红色),即18个未使用的样本和42个用于分析的腺瘤样本无花果1,2,3.,4,6而且7.(e) ROC为测试集。AUC为96%,95% CI为87.88-100%。(f- - - - - -j)训练和测试从对照中分类腺瘤的模型,执行如下一个- - - - - -e.训练集的AUC (n=55个对照组,42个腺瘤)为87.38%,95% CI为80.21-94.55%;测试集(8个对照,5个晚期腺瘤和46个癌)的AUC为59.56%,95% CI为37.51-81.61%。(k- - - - - -o)训练和测试从对照中分类腺瘤的模型,执行如下(f- - - - - -j),除了年龄和BMI与mlg一起被包括在内。年龄由模型选择,使最佳标记数量为11。训练集的AUC为89.74%,95% CI为83.32-96.16%;测试集的AUC为59.56%,95% CI为37.64-81.48%。
在MLG标记中,可能是口服厌氧菌的MLG -75和MLG -84,前者也显示出较高的腺瘤优势比(补充数据5),表明其在发病机制中的早期作用。包括其他MLG标记物拟杆菌massiliensis、mlg-2985、mlg-121及十多个分类上未定义的mlg (补充数据5).因此,癌分类器选择的mlg捕获了腺瘤和癌中不断恶化的肠道微生物组的重要特征,并在这些肿瘤的早期和非侵入性诊断中具有巨大潜力。
我们直接研究了肠道MLGs用于识别腺瘤的用途,腺瘤比结直肠癌更难筛查,但对早期干预很重要3.,23.在5次重复的10倍交叉验证后,随机森林模型选择了10个mlg,允许对训练集进行最佳分类(55个对照组和42个晚期腺瘤;图5 f-h,补充数据1、5).在测试集中(8个对照组,5个晚期腺瘤和46个癌),所有晚期腺瘤样本都被正确分类,而在对照组和癌样本上的表现不尽如人意(被认为是腺瘤阳性的癌,图5 i, j,补充数据1).当年龄和BMI与126个mlg一起被包括在内时,年龄与10个mlg一起被选择为一个标记(图5 km),但在测试集上的表现并没有改善(图5 n, o).因此,粪便mlg为结直肠腺瘤和癌的无创检测提供了新的机会,但确认腺瘤可能需要额外的检查。
与饮食相关的微生物群功能变化
膳食成分如红肉是已知的结直肠癌的危险因素3.,14,15但目前还不清楚饮食是如何对与大肠癌相关甚至导致大肠癌的肠道微生物产生影响的。我们评估了一些临床或生活方式因素对肠道微生物基因或mlg的影响,发现对照、腺瘤或癌状态确实是最强的因素之一(补充图7a,补充数据6).有趣的是,在典型坐标分析(CCA)中,水果和蔬菜消费的影响指向对照富集的MLGs,而c反应蛋白(CRP)和肉类消费与癌富集的MLGs相关(补充图7a).mlg的相对丰度与饲粮或生理参数的Spearman相关系数≥0.2 (图6,补充图8和图9).通过氨基酸发酵产生短链脂肪酸(结肠子的主要能量来源)的富癌细菌和/或代谢胆汁酸的细菌24,25,例如,b . massiliensis,b . dorei,b .通行的,Parabacteroides merdae,答:finegoldii而且b . wadsworthia,与红肉消费量呈正相关,及/或与水果及蔬菜消费量呈负相关(图6),提示结直肠肿瘤发生的共同途径。对照丰富的mlg变异链球菌而且梭状芽胞杆菌Sp .与蔬菜摄入量呈正相关。这些与饮食的弱相关性被高摄入量组和低摄入量组之间mlg的显著差异所支持(补充图7b-h).富癌细菌如b . massiliensis,p . merdae,答:finegoldii而且b . wadsworthia摄入更多蔬菜或水果的受试者是否比摄入更多蔬菜或水果的对照组含量更低变异链球菌而且梭状芽胞杆菌sp。补充图7b,d).在食用大量肉类或红肉的受试者中,包括mlg-84、mg -850和mg -1738在内的癌mlg-84、mg -850和mg -1738更为丰富(补充图7e,f).

计算每个MLG(>100个基因)的相对丰度与危险因素或临床表型水平之间的Spearman相关系数。红色,正相关;蓝色代表负相关。+,P罗斯福= 0.4131 < 0.05;*,P< 0.01,富兰克林·德兰诺·罗斯福= 0.2635。每个MLG的名称根据其富集方向被着色,即,如果对照组比癌组高,则为绿色,如果癌组比对照组高,则为红色。放线菌viscosus,Alistipes putredinis,Paraprevotella克拉拉、mg -703、mg -711、mg -798、mg -1602和mg -7070与26种数值表型均无显著相关性(P>0.05, Spearman’s),未显示。γ -谷氨酰转移酶(GGT)、体重指数(BMI)、丙氨酸转氨酶(ALT)、天冬氨酸转氨酶(AST)、甘油三酯(TG)、血红蛋白(Hb)、低密度脂蛋白(LDL)、c反应蛋白(CRP)、腰臀比(WHR)、高密度脂蛋白(HDL)。
铁蛋白是一种负责细胞内铁储存的蛋白质,血清铁蛋白水平与许多富癌mlg呈负相关(图6),强调铁是许多致病菌生长的关键资源26它们以宿主或肉类等食物来源的铁为食。血红蛋白(Hb)与肿瘤富集的mlg-75、mg -2985、mg -88、mg -84呈负相关。
其他已知的危险因素,如目前或曾经吸烟也与mlg的富集(包括b . dorei而且b . vulgatus;补充图8和图9).腰臀比与对照组为负相关梭状芽胞杆菌sp.和美国酸奶,与癌富集呈正相关拟杆菌Sp ., mg -368及mg -448 (图6).另一方面,BMI与部分富癌mlg呈负相关。这些结果与荟萃分析一致,表明中心型肥胖是比一般肥胖更可靠的CRC危险因素27.
与饮食所起的重要作用一致,磷酸转移酶系统(许多不同糖的转运体)的KEGG orthology (KO)模块在健康对照中与腺瘤样本相比或在腺瘤中与癌样本相比(图7,补充数据7).与腺瘤相比,癌中转运组氨酸、精氨酸和赖氨酸的模块富集,而合成组氨酸、赖氨酸、蛋氨酸、半胱氨酸、亮氨酸和色氨酸的模块在对照组中富集于腺瘤,或腺瘤中富集于癌样本。在腺瘤-癌序列中,除了增加了对膳食氨基酸或宿主氨基酸的利用能力外,还增加了对宿主聚糖(如黏蛋白和糖胺聚糖)的代谢能力,这表明KO模块的丰度较高,可降解硫酸皮聚糖、硫酸肝素和硫酸角蛋白(图7).这些模块中的硫酸酯酶已在黄杆菌属肝素,亚种在其他地方也能看到拟杆菌28,29,30.,31.与对照组相比,腺瘤中的磺酸盐/硝酸盐/牛磺酸转运系统升高,提示胆汁酸代谢发生变化(图7,补充数据7).与健康对照组相比,在腺瘤或癌中也观察到更高水平的甲烷生成模块。此外,这些差异富集的功能,如脂多糖(LPS)生物合成、硫酸盐角蛋白降解和铁(III)转运系统,可以在腺瘤或癌分类器中的MLG标记物中发现,以及更多的室内功能(图7,补充数据8-10).总之,我们的研究结果表明,相对于肉类,低水果和蔬菜的饮食选择了腐败细菌的生长,这可能有助于促进结直肠癌。

比较对照组和腺瘤、腺瘤和癌、对照组和癌之间KO模块的相对丰度,以及报告得分显著差异的模块(< - 1.7,前者富集;>1.7,富集于后者)。+,记者评分>2.3或< - 2.3。
除了KEGG数据库中列出的功能外,据报道肠道菌群还可以控制对癌症治疗的反应6,32.Alistipes而且瘤胃球菌属MC38结肠癌移植C57Bl/6小鼠抗il - 10r /CpG寡核苷酸免疫治疗后TNF生成呈正相关6.我们观察到答:finegoldii, mlg-482(与答:onderdonkii),答:putredinis在大肠癌中,mlg-6514的减少(最与瘤胃球菌属sp. 5_1_39BFAA),其丰度与少数富癌MLGs (图4 b, c,补充数据5).所有这些mlg在接近100%的癌症患者中都存在,无论肿瘤的组织学、分期或位置如何。p . distasonis单关联已被证明会损害阿霉素对小鼠已建立的MCA205肉瘤的免疫原性化疗32.mlg为p . distasonis而且p . merdae在癌中比在晚期腺瘤样本中更丰富,并且在大多数癌样本中检测到(图4 b, c,补充数据5).总的来说,这些结果表明,人类大肠癌中存在或过度生长的肠道微生物可能促进或废除免疫或化疗,并且应该对每个患者进行最佳治疗方案的选择进行检查。
讨论
总之,我们对健康对照组、结直肠腺瘤和癌患者肠道微生物组的宏基因组全关联研究确定了与肿瘤相关的基因、菌株(MLGs)和功能,并为结直肠腺瘤和癌的早期检测和患者分层开辟了新途径。我们的标记物如何在世界各地更大的人群中帮助改善结直肠肿瘤的非侵入性筛查仍有待观察。
在结肠炎相关的CRC小鼠模型中,肠毒素b . fragilis通过T辅助性17型(Th17)炎症反应和粘附-侵袭性反应诱导结肠炎和结肠肿瘤大肠杆菌也会促进癌症33,34,35.b . ovatus而且b . vulgatus在人类克罗恩病病例中(6对不一致双胞胎和4对一致双胞胎)36.的显著增加b . dorei而且b . massiliensis从健康腺瘤到晚期,均显著增加b . massiliensis,b . ovatus,b . vulgatus而且大肠杆菌由晚期腺瘤至癌(图4).b . dorei,b . vulgatus而且大肠杆菌也与c反应蛋白水平相关,c反应蛋白是急性炎症的标志(图6).这些结果表明肠道微生物在结肠炎相关和腺瘤相关的CRC中发挥了类似的作用。
Akkermansia是一种粘液降解细菌,属于疣微菌门据报道,在人类和小鼠模型中与CRC相关37,38.我们观察到在丰度上没有差异Akkermansia在健康对照组中,晚期腺瘤和癌样本(补充图2).三种基于pam的肠型中有两种含有较高水平的Akkermansia,其中分别包括更多的对照组和癌样本(补充图1).考虑到肥胖、饮食和用餐时间等因素的未来分析将有助于解决这种重要细菌在结直肠癌中的可能作用。
即使腐烂的细菌如Alistipes而且拟杆菌氨基酸能产生短链脂肪酸,碳水化合物发酵还是首选24,39,这可能解释了水果和蔬菜的保护作用。在一些梭菌属然而,糖的运输取决于氨基酸的发酵(Glu, Lys, His或Ser)40,41这表明它们只有在氨基酸供应充足的情况下才能茁壮成长。酚类化合物是由芳香族氨基酸苯丙氨酸和酪氨酸发酵产生的39这可能会增加结肠中的DNA损伤。胆汁酸代谢拟杆菌物种和b . wadsworthia也会影响肠道微生物组成并影响宿主生理机能吗42,43.b . wadsworthia特别是利用牛磺酸偶联胆汁酸还原亚硫酸盐,并促进遗传易感小鼠的结肠炎(Il10−−/)44.胆汁酸也被证明能引起DNA损伤并促进小鼠的肝细胞癌45,46.未来的研究将有助于阐明饮食、肥胖和吸烟等已知风险因素如何在结直肠癌的发展过程中共同作用于肠道微生物组。
在对照富集的mlg中有乳酸菌双歧杆菌animalis,变异链球菌而且美国酸奶.产生的乳酸可能有助于降低pH值,抑制氨基酸在结肠中的降解24,39.乳酸菌而且双歧杆菌属刺激NADPH氧化酶1依赖的ROS生成和肠道干细胞增殖47,乳酸盐被报道加速饥饿小鼠结肠上皮细胞的周转48.因此,晚期结直肠腺瘤或癌患者似乎缺乏乳酸生产共生物,如双歧杆菌属这可以促进结肠上皮细胞的日常更新,并抑制潜在的病原体。肠道菌群依赖的饮食或生活方式干预治疗大肠癌值得进一步研究。
方法
研究队列和患者信息
这项研究是根据国家CRC筛查建议对健康筛查计划的参与者进行的49以及2010年至2012年期间在奥伯恩多夫医院(奥地利萨尔茨堡Paracelsus医科大学教学医院)内科进行结肠镜检查的疑似结直肠癌患者的临床检查。该研究得到了当地伦理委员会的批准(Ethikkommission des Landes Salzburg,批准号为;415-E/1262/2-2010)和所有参与者的知情同意。
最初分析包括147名年龄在45-86岁之间的白种人的数据,包括57名健康对照(24名女性和33名男性),44名晚期腺瘤(22名女性和22名男性)和46名癌(18名女性和28名男性)(补充数据1).另外9个样本用于另一份手稿(6个健康对照和3个晚期腺瘤样本,补充数据1)也用于基于mlg的腺瘤或癌分类器的测试集(图5).到目前为止,还没有研究以可比的方式调查给定的主题;因此,无法进行正式的功率分析来计算样本量。然而,从之前对疾病中粪便微生物群的16S和宏基因组短枪测序研究来看,这是一个合理的样本量。受试者按性别、年龄和BMI进行分层,使三组(对照组、晚期腺瘤组和癌组)在这些变量方面具有可比性。晚期腺瘤组14例位于右结肠(包括盲肠、升结肠和横结肠),15例位于左结肠(从脾屈曲到乙状结肠),15例位于直肠。癌组8个位于右结肠,11个位于左结肠,27个位于直肠。结直肠癌按美国癌症联合委员会(AJCC) TNM分期系统分类22.
代谢综合征的评估由国家胆固醇教育计划成人治疗小组定义50.
由护士坐位休息5分钟后测量两次血压,取平均值作为血压测量值。在受试者站直的情况下,在髂骨最高点处测量腰围。当满足以下三项标准时,诊断为代谢综合征:空腹血糖水平≥6.1 mmol−1腰围>102 cm或>88 cm,男性或女性,血压≥130/85 mmhg或目前正在接受降压治疗,血浆甘油三酯≥1.7 mmol l−1,血浆HDL <1.0 mmol l−1或<1.3 mmol l−1男性或女性,或目前的他汀类药物治疗。BMI计算公式为体重/身高平方(kg m−2).
使用Architect CI 4100分析仪,采用铁蛋白化学发光微粒法(ABBOTT Laboratories, ABBOTT Europe, Delkenheim, Germany)测定血清铁蛋白。
实验室评估
在一夜禁食后,所有受试者都获得了静脉血样本,并使用标准的实验室方法进行分析。血液离心,血浆分析甘油三酯,胆固醇,高密度和低密度脂蛋白胆固醇和CRP。在300毫升水中加入75克葡萄糖,进行标准化口服葡萄糖耐量试验。采用Adamts H-8160 (Menarini, Florence, Italy)高效液相色谱法测定HbA1c。稳态模型评估(HOMA-IR;空腹胰岛素(μU l−1) ×空腹血糖(mmol dl−1)/22.5)用于评估胰岛素抵抗。2型糖尿病分为使用糖尿病药物或Hba1C≥6.5%或口服糖耐量试验>11.1 mmol l−12小时后或空腹血糖>7.0 mmol l−1.
粪便样本
收集所有患者和受试者的新鲜粪便样本。用无菌抹刀对样品进行机械均质,然后使用Sarstedt粪便采样系统(Sarstedt, Nümbrecht,德国)取四份。每个样品在一个无菌的12毫升冷冻瓶中含有1克粪便。然后将粪便等分储存在−20°C的家用冰柜中,并在收集后48小时内用冷冻袋装运至实验室,在−80°C下立即储存。患者和受试者在最近3个月内未使用益生菌或抗生素。
结肠镜检查
Klean-Prep泻药(含聚乙二醇59.0 g,硫酸钠5.68 g,碳酸氢钠1.68 g, NaCl 1.46 g,氯化钾0.74 g;Norgine, Marburg, Germany)用于结肠镜检查前的肠道准备。结肠镜检查结果经宏观和组织学结合分析,分为管状腺瘤、晚期腺瘤,即具有绒毛状或管状绒毛状特征,大小≥1cm,或高度发育不良或癌51,52.病变按部位分类(即右结肠包括盲肠、升结肠和横结肠,左结肠包括脾屈至乙状结肠和直肠)。
生活方式和饮食习惯的评估
详细的病史,包括生活方式和饮食问卷。吸烟状况分为从不吸烟、曾经吸烟和现在吸烟(包括每天对现在和以前吸烟的详细评估;每年以包报告的数据)。使用国际身体活动问卷(IPAQ)评估身体活动53根据公布的评分方案,受试者被分为三组:低强度、中等强度和高强度体育活动。在粪便捐献和结肠镜检查后1周内,使用详细的标准化问卷评估饮食习惯。每份的份量及纤维含量是根据美国心脏协会(www.heart.org).肉类消费被详细询问了猪肉、牛肉、小牛肉和鹿肉(归为红肉);鸡肉和火鸡(白肉)和内脏。此外,还评估了蔬菜、水果和鱼类的消费频率和数量,并计算了纤维的总摄入量。
该研究得到了当地伦理委员会的批准(Ethikkommission des Landes Salzburg,批准号为;415-E/1262/2-2010)和所有参与者的知情同意。
宏基因组测序与基因目录构建
在Illumina平台上进行双端宏基因组测序(插入物大小350 bp, reads长度100 bp),测序reads进行质量控制和测序新创使用SOAPdenovo v2.04组装成contigs(参考16,54;除-K 51 -M 3 -F -u外的默认参数)。
使用GeneMark v2.7d对组装的contigs进行基因预测。用BLAT去除冗余基因55以90%重叠和95%同一性为截断点(不允许有间隙)。基因的相对丰度是通过使用与参考文献相同的程序将高质量的测序reads与基因目录对齐来确定的。16.
利用内部管道根据IMG数据库(v400)对预测基因进行分类赋值16(BLASTN v2.2.24, -e 0.01 -b 100 - k1 -F T -m 8),重叠80%,同源性前10%得分为65%。若仍有多个同源性,则该分类单元的同源性为65%,属同源性为85%,种同源性为95%,一致性≥50%。
稀疏曲线
进行稀疏分析以评估健康对照、晚期腺瘤和癌样本的基因丰富度。对于给定数量的样本,我们在队列中进行100次随机抽样,并通过Chao2丰富度估计器估计可以从这些样本中识别出的基因总数56.为了尽量减少错误识别,只有≥1对映射reads的基因被确定存在于样本中。
毒力因子的量化
假定的氨基酸序列与致病细菌毒力因子数据库(VFDB)中的蛋白质进行比对。18使用BLASTP (v2.2.24,除-p BLASTP -a 2 -F F -e 1e-3 -m 8外的默认参数)。通过包含标识>35%和高分片段对评分>60位的最高评分注释命中,将蛋白质分配到毒性因子。采用Kruskal-Wallis试验鉴定差异富集的毒力因子。
微生物群落类型(肠型)
利用属的相对丰度,采用基于pam的方法分析各粪便宏基因组样本的群落类型16,19,并采用基于Dirichlet多项混合模型的测序reads计数方法20.(补充的方法).
全宏基因组关联研究(MGWAS)
为了比较健康对照组、晚期腺瘤和癌患者的粪便微生物群,我们确定了两组中任何一组之间相对丰度有显著差异的基因(Benjamin-Hochberg问-value<0.1, Kruskal-Wallis检验)。然后,根据这些标记基因在所有三组样本中的丰度变化,将它们聚成mlg16.147个样本中有9个含有20%的>埃希氏杆菌属(2个对照,2个腺瘤和5个癌样本),随后仅用于基于mlg的腺瘤或癌分类器的测试集(图5).另外9个样本用于另一份手稿(6个健康对照和3个晚期腺瘤样本,补充数据1)也用于分类器的测试集。
如前所述,根据分类及其组成基因的相对丰度对mlg进行分类学分配和丰度分析16.简单地说,分配到物种需要一个MLG中>90%的基因与物种基因组一致,>95%的同源性和70%的查询重叠。将一个MLG指定为一个属需要>80%的基因与DNA和蛋白质序列中85%相同的基因组一致。在对照组和腺瘤两组比较时,根据mlg在所有对照组和腺瘤样本中丰度的Spearman相关性进一步聚类,并通过Cytoscape 3.0.2显示共现网络。富集方向采用Wilcoxon秩和检验(P< 0.05)。
MLG-based分类器
在随机森林模型(R 3.0.2, randomForest4.6-7包)上使用对照、晚期腺瘤或癌样本(补充的方法).对10次交叉验证的5次试验的交叉验证误差曲线(平均每组10个测试集)进行平均,并将平均曲线中的最小误差加上该点的s.d.作为截止点。列出误差小于截止值的所有MLG标记集(≤50个),选择MLG数量最少的集合作为最优集合。使用这组mlg计算腺瘤或癌的概率,并绘制ROC (R 3.0.2, pROC3包)。在测试集上对模型进行进一步检验,确定预测误差。
PERMANOVA对临床和生活方式因素的影响
置换多元方差分析(PERMANOVA)57对所有样本的基因丰度谱进行分析,以评估所列每个因素的影响(补充的方法).我们在R(3.0.2,纯素包)中使用了欧几里得距离和9999个排列58).
典型对应分析
对对照、腺瘤和癌样本的MLG(>100个基因)丰度谱进行典型对应分析,以评估所列每个因素的影响(补充的方法).图由R(3.0.2,素食包58).
KEGG分析
假定的氨基酸序列从基因目录中翻译出来,并使用BLASTP (v2.2.24,除-e 0.01 -b 100 - k1 -F T -m 8外的默认参数)与KEGG数据库中的蛋白质/结构域(版本59.0,去除动植物基因)进行比对。每个蛋白质通过包含至少一个HSP评分>60位的最高评分注释命中(s)分配到KO组。
差异富集KO模块根据其报告评分进行识别59从Z-个人ko的分数。对5个以上样本中出现的所有ko进行单尾Wilcoxon秩和检验,并使用Benjamin-Hochberg程序进行多次测试调整。的Z-可以计算每个KO的得分:
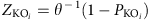
在哪里θ−1是逆正态累积分布,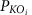 调整后的PKO的价值。聚合Z- KEGG路径(或模块)的得分是:
调整后的PKO的价值。聚合Z- KEGG路径(或模块)的得分是:
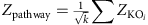
在哪里k为路径(或模块)中涉及的ko数量。
的背景分布Z通路减去平均值(μk),再除以s.d。(σk)Z-得分1000套kKO,从整个代谢KO网络中随机选择:
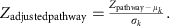 的Zadjustedpathway作为评估特定通路或模块富集程度的最终报告评分。报告评分≥1.6(符合正态分布的置信度为90%)可作为显著鉴别通路的检测阈值。
的Zadjustedpathway作为评估特定通路或模块富集程度的最终报告评分。报告评分≥1.6(符合正态分布的置信度为90%)可作为显著鉴别通路的检测阈值。
额外的信息
加入代码:所有156个样本的宏基因组霰弹枪测序数据已存入EBI数据库,登录代码为ERP008729。
如何引用这篇文章:冯,Q。等.肠道微生物组沿结直肠腺瘤-癌序列的发展。Commun Nat。6:6528 doi: 10.1038/ncomms7528(2015)。
参考文献
杰马尔等人。全球癌症统计。CA Cancer J. Clin。61, 69-90(2011)。
Ferlay, J.等人。GLOBOCAN 2012 v1.0,全球癌症发病率和死亡率:国际癌症研究机构癌症基地第11号国际癌症研究机构(2013)http://globocan.iarc.fr.
布伦纳,H., Kloor, M. & Pox, C. P.结直肠癌。《柳叶刀》383, 1490-1502(2014)。
Vogelstein, B. & Kinzler, K. W.癌症基因及其控制途径。Nat,地中海。10, 789-799(2004)。
格里文尼科夫,s.i.等人。腺瘤相关屏障缺陷和微生物产物驱动IL-23/ il -17介导的肿瘤生长。自然491, 254-258(2012)。
Iida, N.等。共生菌控制肿瘤微环境。科学342, 967-970(2013)。
Belcheva, A.等人。肠道微生物代谢驱动缺乏msh2的结肠上皮细胞的转化。细胞158, 288-299(2014)。
Kostic, a.d.等人。基因组分析确定梭杆菌与结直肠癌的相关性。基因组Res。22, 292-298(2012)。
Castellarin, M.等。有核梭杆菌感染在人大肠癌中普遍存在。基因组Res。22, 299-306(2012)。
Gevers, D.等人。新发克罗恩病中未治疗的微生物群。细胞宿主微生物15, 382-392(2014)。
Kostic, a.d.等人。有核梭杆菌增强肠道肿瘤发生和调节肿瘤免疫微环境。细胞宿主微生物14, 207-215(2013)。
秦,等。宏基因组测序建立的人体肠道微生物基因目录。自然464, 59-65(2010)。
李,J.等。人类肠道微生物组内参基因的综合目录。生物科技Nat。》。32, 834-841(2014)。
对乳腺癌和结肠癌病因的研究。自然338, 389-394(1989)。
杨,K.等。饮食对散发性结肠癌小鼠模型结肠肿瘤的诱导作用。癌症Res。68, 7803-7810(2008)。
秦,等。2型糖尿病患者肠道菌群宏基因组范围的关联研究。自然490, 55-60(2012)。
Sanapareddy, N.等人。直肠微生物丰富度的增加与人类结直肠腺瘤的存在有关。ISME J。6, 1858-1868(2012)。
陈丽丽,熊志,孙丽丽,杨娟,金强。VFDB 2012年最新进展:细菌毒力因子的遗传多样性和分子进化。核酸测定。40, d641-d645(2012)。
Arumugam等人。人类肠道微生物群的肠型。自然473, 174-180(2011)。
Ding, T. & Schloss, P. D.人体微生物群落类型的动力学和关联。自然509, 357-360(2014)。
骑士,D.等人。反思“欠佳”。细胞宿主微生物16, 433-437(2014)。
目前结直肠癌的TNM分期。柳叶刀杂志。8, 572-573(2007)。
Imperiale, t.f.等。多靶点粪便DNA检测用于结直肠癌筛查。心血管病。j .地中海。370, 1287-1297(2014)。
Smith, E. & Macfarlane, G.人类大肠中氨基酸发酵细菌的计数:pH值和淀粉对肽代谢和氨基酸异化的影响。《。生态。25, 355-368(1998)。
Narushima, S.等。与人类肠道细菌相关的去氧胆酸在无菌小鼠中的形成。脂质41, 835-843(2006)。
贾基,T.等人。在肯尼亚婴儿中,铁的强化会对肠道微生物组产生不利影响,增加病原体丰度并诱发肠道炎症。肠道pii, gutjnl-2014-307720(2014)。
Ma, Y.等人。肥胖与结直肠癌风险:前瞻性研究的系统回顾。《公共科学图书馆•综合》8, e53916(2013)。
Hwa, V. & Salyers, A. A.两种利用硫酸软骨素的拟杆菌(Bacteroides thetaotaomicron)突变体的分析,它们在无菌小鼠胃肠道中与野生型竞争的能力不同。达成。环绕。Microbiol。58, 869-876(1992)。
拉曼,R.等。肝素黄杆菌的肝素/硫酸肝素2- o -磺化酶。酶活性位点和糖底物特异性的结构和生化研究。生物。化学。278, 12167-12174(2003)。
米耶特,J. R.等。肝素黄杆菌的肝素/硫酸肝素2- o -磺化酶。分子克隆、重组表达及生化表征。生物。化学。278, 12157-12166(2003)。
乌尔默,j.e.等。来自人类肠道共生拟杆菌taotaomicron的糖胺聚糖(GAG)硫酸酯酶的特征揭示了第一个GAG特异性细菌硫酸酯酶。生物。化学。289, 24289-24303(2014)。
Viaud, S.等人。肠道菌群调节环磷酰胺的抗癌免疫作用。科学342, 971-976(2013)。
吴,S.等。人类结肠共生体通过激活辅助性17T细胞反应促进结肠肿瘤的发生。Nat,地中海。15, 1016-1022(2009)。
亚瑟,J. C.等。肠道炎症的目标是微生物群的致癌活性。科学338, 120-123(2012)。
亚瑟,J. C.等。微生物基因组分析揭示炎症在细菌诱导的结直肠癌中的重要作用。Commun Nat。5, 4724(2014)。
Dicksved, J.等人。克罗恩病同卵双胞胎肠道菌群的分子分析。ISME J。2, 716-727(2008)。
Baxter, n.t., Zackular, J. P., Chen, g.y.和Schloss, P. D.在人类粪便定殖后肠道微生物组的结构决定结肠肿瘤负荷。微生物组2, 20(2014)。
Weir, t.l.等。结直肠癌患者与健康成人粪便微生物组和代谢组的差异《公共科学图书馆•综合》8, e70803(2013)。
Smith, E. A. & Macfarlane, G. T.人类结肠细菌产生酚类和吲哚类化合物的计数:pH值、碳水化合物可用性和保留时间对异种芳香族氨基酸代谢的影响。j:。Bacteriol。81, 288-302(1996)。
Robrish, S. A., Oliver, C. & Thompson, J.梭杆菌的糖代谢:转运、磷酸化的调节,以及死亡梭杆菌ATCC 25557聚合物的形成。感染。Immun。59, 4547-4554(1991)。
Robrish, S. A., Oliver, C. & Thompson, J.核酸梭杆菌ATCC 10953对糖的氨基酸依赖转运。j . Bacteriol。169, 3891-3897(1987)。
伊斯兰教,K. B.等。胆汁酸是调节大鼠盲肠菌群组成的宿主因子。胃肠病学141, 1773-1781(2011)。
萨因,s.i.等人。肠道菌群通过降低牛磺酸- β -胆酸(一种天然存在的FXR拮抗剂)的水平来调节胆汁酸代谢。细胞。金属底座。17, 225-235(2013)。
Devkota, S.等人。膳食脂肪诱导的牛磺胆酸促进Il10-/-小鼠的病原菌扩张和结肠炎。自然487, 104-108(2012)。
杨,F.等。胆汁酸受体法尼索X受体缺失时肝脏肿瘤的自发发展。癌症Res。67, 863-867(2007)。
Yoshimoto, S.等人。肥胖诱导的肠道微生物代谢产物通过衰老分泌体促进肝癌的发生。自然499, 97-101(2013)。
琼斯,R. M.等。共生乳酸菌通过nox介导的活性氧产生刺激肠道上皮细胞增殖。EMBO J。32, 3017-3028(2013)。
冈田,T.等。微生物源性乳酸加速饥饿小鼠结肠上皮细胞的转化。Commun Nat。4, 1654(2013)。
斯塔德迈尔,A.等人。非酒精性脂肪肝:结直肠瘤变的独立危险因素j .实习生。地中海。270, 41-49(2011)。
成人高血胆固醇检测和治疗专家小组,E.国家胆固醇教育计划(NCEP)成人高血胆固醇检测、评估和治疗专家小组(成人治疗小组III)第三次报告执行摘要。j。地中海,协会。285, 2486-2497(2001)。
结直肠息肉指南:结直肠息肉患者的诊断、治疗和监测点。j .杂志。95, 3053-3063(2000)。
Winawer, s.j. & AG。,Z.的一个dvanced adenoma as the primary target of screening.Gastrointest。Endosc。中国。n。12, 1-9(2002)。
克雷格,c.l.等。国际体育活动问卷:12国信度和效度。医学科学。体育Exerc。35, 1381-1395(2003)。
Luo, R.等。SOAPdenovo2:经验改进的内存效率短读从头汇编程序。Gigascience1, 18(2012)。
Kent, W. J. blat -类似blast的对齐工具。基因组Res。12, 656-664(2002)。
Chao, A.估计捕获-再捕获数据在不平等可捕获性下的种群规模。生物识别技术43, 783-791(1987)。
麦卡德尔,B. H. &安德森,M. J.拟合多元模型到社区数据:对基于距离的冗余分析的评论。生态82, 290-297(2001)。
Zapala, m.a. & Schork, N. J.距离矩阵的多元回归分析,用于测试基因表达模式和相关变量之间的关联。国家科学院学报美国103, 19430-19435(2006)。
杨晓明,李志强,李志强,等。利用代谢网络拓扑结构揭示代谢的转录调控。国家科学院学报美国102, 2685-2689(2005)。
确认
本研究得到中国自然科学基金(30890032,30725008,30811130531和31161130357),中国深圳市政府(BGI20100001, CXB201108250096A和CXB201108250098A),丹麦战略研究委员会资助(2106-07-0021),丹麦自然科学研究委员会Ole RØmer资助和Solexa项目(272-07-0196)的支持。我们感谢奥伯恩多夫医院的Elke Albrecht、Monika Ratkowitsch和Carmen Winkler,以及深圳华大基因的同事们在DNA提取、文库建设、测序和讨论方面的技术支持。
作者信息
作者及隶属关系
贡献
其子as U.H.-S。和C.D.设计并实施了临床研究。h.t., C.D.和juw。发起并指导项目。Q.F、S.L l、h.j.、L.T t、Z.L l、j.l l和L.X进行了生物信息学分析并制备了图。所有作者都对手稿的撰写和修改做出了贡献。
相应的作者
道德声明
相互竞争的利益
作者声明没有相互竞争的经济利益。
补充信息
补充信息
补充图1-9,补充方法及补充参考资料(PDF 274kb)
补充数据集1
所有样本的临床资料。(xlsx61 kb)
补充数据集2
为每个样本生成测序数据。(xlsx21 kb)
补充数据集3
毒力因子在健康、晚期腺瘤和癌样本中差异富集。根据毒力因子数据库(VFDB)。(xlsx27 kb)
补充数据集4
三组间表型的一致性。(xlsx11kb)
补充数据集5
包含超过100个基因的所有mlg的丰度、注释和优势比。富集方向采用Wilcoxon秩和检验,p< 0.05, Bonferroni校正为对照进行多次试验。(xlsx59 kb)
补充数据集6
临床记录或日常生活习惯对肠道微生物基因谱的影响。(xlsx12kb)
补充数据集7
在健康、晚期腺瘤和癌样本中差异富集的KO通路。(xlsx17 kb)
补充数据集8
在腺瘤或癌分类器中使用的MLG标记中发现KO模块。(xlsx52 kb)
补充数据集9
在腺瘤或癌分类器中使用的MLG标记物中发现KO通路。(xlsx34kb)
补充数据集10
基于mlg的分类器中KO模块和路径的总结。只要可以识别出一个或多个模块或路径,就认为它存在于MLG中。(xlsx20 kb)
权利和权限
关于本文

引用本文
冯强,梁硕,贾宏,李志强,李志强。et al。肠道微生物组沿结直肠腺瘤-癌序列的发展。Nat Commun6, 6528(2015)。https://doi.org/10.1038/ncomms7528
收到了:
接受:
发表:
DOI:https://doi.org/10.1038/ncomms7528
进一步的阅读
宿主-微生物组蛋白质-蛋白质相互作用捕获疾病相关途径
基因组生物学(2022)
人类肠道微生物组中硫代谢基因的多样性和分布及其与结直肠癌的关系
微生物组(2022)
代谢:微生物基因组的功能性状、代谢、生物地球化学和群落规模的功能网络的高通量分析
微生物组(2022)
结直肠癌患者微生物群多样性和组成的全营养相关性研究
BMC癌症(2022)
中国根头菌中肠微生物组的宏基因组学研究
寄生虫和病媒(2022)
