摘要
背景
RNA-Seq正在革新测量转录物丰度的方法。从RNA-Seq数据进行转录物量化的一个关键挑战是如何处理映射到多个基因或异构体的读取。在没有测序基因组的情况下,这个问题对于用新生转录组组合进行量化尤其重要,因为很难确定哪些转录本是同一基因的同工型。第二个重要的问题是RNA-Seq实验的设计,包括读取次数、读取长度以及读取是来自cDNA片段的一端还是两端。
结果
我们提出了RSEM,一个用户友好的软件包,用于从单端或成对端RNA-Seq数据中量化基因和亚型丰度。RSEM输出丰度估计、95%可信区间和可视化文件,还可以模拟RNA-Seq数据。与其他现有工具相比,该软件不需要参考基因组。因此,与全新的转录组组装器结合,RSEM能够对没有测序基因组的物种进行准确的转录本定量。在模拟和真实的数据集上,RSEM比依赖参考基因组的量化方法具有更好或相当的性能。利用RSEM有效使用模糊映射读取的能力,我们表明,精确的基因水平丰度估计最好通过大量短的单端读取获得。另一方面,根据每个基因可能的剪接形式的数量,通过使用配对端读取可以提高单个基因中异构体的相对频率的估计。
结论
RSEM是一种精确和用户友好的软件工具,用于从RNA-Seq数据量化转录丰度。由于它不依赖于参考基因组的存在,因此它对新生转录组集合的量化特别有用。此外,RSEM为RNA-Seq定量实验的成本效益设计提供了有价值的指导,目前这是相对昂贵的。
背景
RNA-Seq是一种分析转录组的强大技术,预计将取代微阵列[1].利用测序技术的最新进展,RNA- seq实验从样本RNA片段衍生的cdna末端产生了数百万个相对较短的reads。产生的reads可用于许多转录组分析,包括转录本定量[2- - - - - -7,差异表达检测[8,9,基于参考的基因注释[6,10]和de novo转录本组合[11,12].在这篇论文中,我们专注于转录本定量的任务,这是估计相对丰度,在基因和亚型水平。测序之后,量化任务通常包括两个步骤:(1)将reads映射到参考基因组或转录本集,(2)基于reads映射估计基因和亚型丰度。
量化的一个主要问题是RNA-Seq reads并不总是唯一地映射到单个基因或亚型。之前,我们已经证明,在统计模型中适当考虑读取映射的不确定性对于实现最准确的丰度估计至关重要[7].在本文中,我们提出了一个用户友好的软件包RSEM (RNA-Seq by Expectation Maximization),它实现了我们的量化方法,并对我们的原始模型进行了扩展。RSEM的一个关键特征是不需要参考基因组。相反,它只需要用户提供一组参考转录本序列,例如由从头转录组组装器生成的转录本序列[11,12].对我们原始方法的扩展包括成对端(PE)和可变长度读取、片段长度分布和质量评分的建模。此外,现在计算每个基因和亚型丰度的95%可信区间(CI)和后验均值估计(PME),以及最大似然估计(ML)。最后,RSEM现在可以通过概率加权读取对齐和读取深度图实现输出的可视化。
通过模拟和真实RNA-Seq数据的实验,我们发现RSEM具有优于或相当于其他相关方法的量化精度。通过额外的实验,我们得到了两个令人惊讶的结果,关于PE数据的价值和评估转录本丰度的质量评分信息。虽然PE读取比单端(SE)读取提供更多的信息,但我们的实验表明,在相同的测序吞吐量(根据测序碱基的数量)下,短SE读取允许在基因水平上最好的量化精度。虽然人们会假设质量分数为正确的读取映射提供了有价值的信息,但我们发现,对于具有类似illumina的错误配置文件的RNA-Seq读取,考虑质量分数的模型并不比只使用读取序列的模型显著提高量化精度。
相关工作
一些最初的RNA-Seq论文中使用的一种简单的量化方法[13,14],今天仍在使用的方法是计算唯一映射到每个基因的读取数,可能通过基因序列的“可映射性”来纠正基因的计数[15,以及它的长度。这种方法的主要问题是:(1)丢弃数据,如果不考虑“可映射性”,就会产生有偏差的估计,(2)对选择性拼接基因的估计不正确[16]和(3)不能很好地扩展到估计异构体丰度的任务。后来开发了一些方法,通过“拯救”映射到多个基因的读取(“多重读取”)来解决第一个问题[17,18].其他一些方法解决了后两个问题,但没有解决第一个问题,方法是在亚型水平对RNA-Seq数据建模[5].后来,我们开发了RSEM背后的方法,该方法通过使用RNA-Seq读取的生成模型和EM算法来估计异构体和基因水平的丰度,解决了所有这些问题[7].自RSEM方法发表以来,人们开发了许多利用类似统计方法的方法[3.,4,6,19- - - - - -22].
在已开发的方法中,只有RSEM和IsoEM能够完全处理异构体和基因之间映射不明确的读数,这两种方法的作者已经表明,这对于实现最佳估计精度非常重要[4,7].与IsoEM相比,RSEM能够建模非均匀序列无关的读起始位置分布(rspd),如一些RNA-Seq协议产生的3'偏分布[1].此外,RSEM可以计算PME和95% ci,而IsoEM只产生ML估计。最后,RSEM是我们所知的唯一一种统计方法,它可以在没有整个基因组序列的情况下工作,这样就可以对只有转录本序列的物种进行RNA-Seq分析。
实现
RSEM的典型运行只包含两个步骤。首先,生成一组参考转录本序列并对其进行预处理,以供后续的RSEM步骤使用。其次,将一组RNA-Seq reads与参考转录本进行对齐,并将得到的对齐结果用于估计丰度及其可信区间。这两个步骤由用户友好的脚本执行rsem-prepare-reference而且rsem-calculate-expression.RSEM工作流的步骤如图所示1并在接下来的部分中进行更详细的描述。

RSEM软件工作流程.标准的RSEM工作流(由实箭头表示)由运行两个程序(rsem-prepare-reference而且rsem-calculate-expression),它自动使用Bowtie进行读取对齐。带有可选对齐程序的工作流还使用由虚线箭头连接的步骤。另外两个项目,rsem-bam2wig而且rsem-plot-model,允许可视化RSEM的输出。RNA-Seq数据也可以通过RSEM通过虚线箭头所示的工作流程进行模拟。
参考序列准备
RSEM被设计用于与转录本序列对齐的reads,而不是全基因组序列。使用转录水平校准有几个优点。首先,对于真核生物样本,RNA-Seq reads到基因组的对齐通过剪接和聚腺苷酸化变得复杂。跨越剪接结或延伸到poly(A)尾的Reads在基因组水平上对齐是具有挑战性的,尽管已有工具可用于对齐剪接结Reads [23- - - - - -25].其次,使用转录水平的校准可以很容易地分析来自没有测序基因组但具有适当特征的转录组的物种的样本(可能通过RNA-Seq转录组组装[11,12])。最后,所有可能的转录本的总长度通常比基因组的长度要小得多,这使得在转录水平上可以更快地对齐。
可以指定一组转录本rsem-prepare-reference有两种方式。最简单的方法是提供一个fasta格式的文本序列文件。例如,这样的文件可以从参考基因组数据库、从头转录组组装器或EST数据库中获得。或者,使用——gtf选项,可提供基因注释文件(GTF格式)和全基因组序列(FASTA格式)。对于经常研究的物种,这些文件可以很容易地从数据库下载,如UCSC基因组浏览器数据库[26和ensemble [27].如果对现有基因注释的质量有疑问,可以使用基于参考的RNA-Seq转录组组装器,如Cufflinks [28],以提供一套改进的GTF格式的基因预测。当需要基因水平丰度估计时,可以指定一个附加文件,指定哪些转录本来自同一基因(通过——transcript-to-gene-map选项),或者,如果提供了一个GTF文件,则可以使用每个转录本的“gene_id”属性来确定基因成员。使用任何一种指定转录本的方法,RSEM都会生成它自己的一组预处理转录本序列,供后面的步骤使用。对于poly(A) mRNA分析,RSEM将添加poly(A)尾部序列到参考转录本,以允许更准确的读取对齐(禁用——no-polyA).每个参考转录组只需要运行一次准备参考序列的脚本,因为转录本序列以样本无关的方式进行预处理。
读取映射和丰度估计
的rsem-calculate-expression脚本处理与参考转录本序列的读取对齐和相对丰度的计算。默认情况下,RSEM使用蝴蝶结对齐程序[29与专门为RNA-Seq量化选择的参数对齐。或者,用户可以手动运行不同的对齐程序,并以SAM格式提供对齐[30.)rsem-calculate-expression.
当使用替代对准器时,必须小心地适当设置对准器参数,以便RSEM可以提供最佳的丰度估计。首先,也是最关键的一点是,对齐器必须配置为报告读取的所有有效对齐,而不仅仅是单个“最佳”对齐。其次,我们建议对对齐器进行配置,以便在确定有效对齐时只考虑每个读取的短前缀(“种子”)内的匹配和不匹配。例如,在默认情况下,RSEM运行Bowtie来查找前25个垒中最多有两个不匹配的读的所有对齐。其思想是允许RSEM决定哪些对齐最有可能是正确的,而不是将此责任交给对齐器。由于RSEM使用了比读对齐器更详细的RNA-Seq读生成过程模型,这导致了更准确的估计。最后,为了减少RSEM的运行时间和内存使用,可以配置对齐器来抑制对大量(例如> 200)有效对齐的读取的对齐报告。
原来的RSEM包只支持固定长度的SE RNA-Seq读取,不支持质量评分信息,而新的包支持各种各样的输入数据类型。RSEM现在支持SE和PE的读取和可变长度的读取。读取可以以FASTA或FASTQ格式给出。如果以FASTQ格式给出读数,RSEM将使用质量评分数据作为其统计模型的一部分。如果没有提供质量评分,RSEM使用我们先前描述的位置相关的错误模型[7].
读取对齐后,RSEM使用其统计模型的期望最大化(EM)算法计算ML丰度估计(见方法)。有许多选项可用于指定RSEM使用的模型,它应该根据产生输入读取的RNA-Seq协议进行定制。例如,如果使用特定于链的协议,则——strand-specific选项。否则,就假定读具有来自义方向和反义方向的同等概率。碎片长度分布由——片段长度选项族,这对SE分析特别重要。对于PE分析,RSEM从数据中学习片段长度分布。如果协议产生高度5'或3'偏置的读位置分布,则——estimate-rspd选项应指定,以便RSEM可以估计读取起始位置分布(RSPD),这可能允许更准确的丰度估计[7].
除了计算ML丰度估计外,RSEM还可以使用其模型的贝叶斯版本来生成每个基因和亚型丰度的PME和95% CI。这些值是通过吉布斯抽样(参见方法)计算出来的,可以通过指定——calc-ci选择。95%的ci对于评估样本间的差异表达是有价值的,特别是对于重复基因或异构体,因为ci捕获了由于随机采样效应和读取映射的模糊性而产生的不确定性。我们建议将ci与差分表达式工具的结果结合使用,目前差分表达式工具没有考虑多读分配的差异。PME值可以用来代替ML估计,因为它们非常相似,但具有通常包含在95% ci内的方便属性,这有时不是小ML估计的情况。
RSEM的主要输出由两个文件组成,一个用于isoform级别的估计,另一个用于基因级别的估计。丰度估计是根据两种方法给出的。第一个是估计从一个给定的亚型或基因中衍生出来的片段的数量。我们只能估计这个数量,因为阅读通常不会唯一地映射到单个文本。该计数通常是一个非整数值,是给定ML丰度的同源型或基因中可对齐的和未过滤的片段数量的期望。这些(可能四舍五入)计数可由微分表达式方法使用,如edgeR [9]或DESeq [8].第二种衡量丰度的方法是估计某一特定亚型或基因所组成的转录本的比例。这个度量可以直接用作0到1之间的值,也可以乘以106以每百万人学习成绩(TPM)计算。转录分数测量优于流行的RPKM [18]及FPKM [6,因为它独立于平均转录本表达长度,因此在样本和物种之间更具可比性[7].
可视化
RSEM可以在基因组浏览器中生成RNA-Seq数据的两种不同可视化输出作为轨迹,如UCSC基因组浏览器[31].当指定——out-bam选项时,RSEM映射将从转录到基因组坐标的对齐读取,并以BAM格式输出结果对齐[30.].BAM文件中的每个对齐(使用MAPQ字段)都根据它是真实对齐的概率进行加权,前提是RSEM学习到的ML参数。基因组浏览器中BAM文件的可视化使用户能够看到所有的读取对齐和RSEM分配给它们的后验概率。方法可以进一步处理BAM文件rsem-bam2wig程序生成UCSC wigg格式的文件,在给定ML参数的情况下,给出每个基因组位置重叠的预期读取数。Wiggle可视化对于查看跨文本的读取分布非常有用。图中显示了UCSC基因组浏览器中BAM和WIG可视化的一个例子2.要产生这两种可视化,必须向参考准备脚本提供一个gtf格式的注释文件,以便读取对齐可以映射回基因组坐标。

UCSC基因组浏览器中的RSEM可视化.UCSC基因组浏览器中小鼠RNA-Seq数据集SRR065546的RSEM输出示例可视化。(A)摆动输出和BAM输出的同步可视化,前者给出基因组中每个位置的预期读取深度,后者给出概率加权读取对齐。在BAM跟踪中,成对的读取由一条细黑线连接,读取的黑暗表示其对齐的后验概率(黑色表示高概率)。(B)期望读深度(顶部轨迹)与唯一映射只读(底部轨迹)计算的读深度有很大差异的示例基因。
为了帮助诊断RNA-Seq数据生成或量化中的潜在问题,RSEM还允许从给定样本中学习到的测序模型的可视化。这是通过运行rsem-plot-model程序上的输出rsem-calculate-expression.许多情节是由rsem-plot-model,包括学习到的片段和读长度分布、RSPD和测序错误参数。为SRA实验SRX018974的RNA-Seq数据集生成的三个图[25]显示在附加文件中1.
模拟
RSEM还允许根据其所基于的生成模型对RNA-Seq数据集进行模拟(参见方法)。模拟是由rsem-simulate-reads程序,该程序采用输入丰度估计、排序模型参数和参考转录本(由rsem-prepare-reference).通常,丰度估计和排序模型是通过在真实数据集上运行RSEM获得的,但它们也可以手动设置。
结果与讨论
与相关工具的比较
为了评价RSEM,我们将其性能与一些相关的量化方法进行了比较。我们比较了IsoEM (v1.0.5) [4],袖扣(v1.0.1) [6], rQuant (v1.0) [2],以及RSEM的原始实现(v0.6) [32].味噌(3.],它使用了类似于RSEM、IsoEM和Cufflinks的概率模型,但没有包括在比较中,因为它目前只计算每个基因的备选剪接形式的相对频率,而不是全局转录片段。为了使比较公平,我们只在Cufflinks的量化模式下运行。也就是说,它被配置为计算我们提供给所有方法的基因注释集的丰度估计,不允许预测新的转录本。袖扣和rQuant都需要对基因组序列的读取进行校准,我们使用了TopHat [24为了这个目的。TopHat提供了基因注释和平均片段长度,不允许预测新的剪接连接。对于需要比对转录本序列的RSEM和IsoEM,我们使用Bowtie [29].由于用于评估RNA-Seq量化方法准确性的“金标准”数据有限,我们在模拟数据和真实数据上对方法进行了测试。在模拟数据上,我们还测量了这些方法的计算性能(在时间和内存方面)。
模拟数据
由于没有发表的RNA-Seq数据模拟器,我们使用RSEM软件包中包含的模拟器进行了实验。这个模拟器使用了简单和广泛使用的RNA-Seq片段的模型,从一个样本中的转录本中所有可能的起始位点进行统一和独立的采样。用于模拟的模型与Cufflinks和IsoEM显式假设的模型相同,而rQuant隐式使用的模型相同。因此,我们的模拟实验是测试当数据是从它们假设的模型生成时,各种方法的表现如何。我们最初尝试使用一个未发布的外部模拟软件包Flux Simulator [33],但软件中的几个bug使我们无法将其用于本文的目的。
我们使用模拟器以非链特异性的方式从小鼠转录组中生成了一组2000万RNA-Seq片段。从这些片段模拟成对端读取,通过简单地抛出每对的第二个读取来构建单端读取集。使用了两个鼠标引用转录集:RefSeq注释[34]和ensemble注释[27(参见方法)。RefSeq是保守的,有20852个基因,平均每个基因1.2亚型。相比之下,ensemble集合有22,329个基因,平均每个基因有3.4个异构体。我们已经在RSEM网站上提供了这个实验的模拟数据。
对于每个模拟集,我们用测试方法计算丰度估计,并使用我们以前使用的中位数误差百分比(MPE)、误差百分比(EF)和假阳性(FP)统计量来测量转录分数估计的准确性[7].MPE是估计值与真实值之间的百分比误差的中位数。10% EF是丰度估计的百分比误差大于10%的转录本的分数。最后,FP统计量是真实丰度小于1个TPM且被预测至少具有1个TPM丰度的转录本的部分。这些统计数据计算了三个层次的估计:(1)基因相对丰度,(2)全局亚型相对丰度,(3)基因内亚型相对丰度。
数字3.给出了五种方法的丰度估计误差在RefSeq模拟集上的分布,使用[引入的绘图风格。4].表格1给出了方法的MPE、10% EF和FP率。对ensemble模拟集的结果显示在附加文件中2.RSEM v0.6和rQuant只在SE数据上运行,因为它们不处理PE数据。

四种RNA-Seq定量方法的准确性.RSEM、IsoEM、Cufflinks和rQuant对模拟RNA-Seq数据的估计误差百分比分布。PE数据中全局亚型和基因估计的误差分布分别见(A)和(B)。SE数据的全局亚型和基因估计误差分布分别如(C)和(D)所示。
对于PE和SE读取,RSEM和IsoEM优于Cufflinks和rQuant。这两对方法之间的性能差距可能有两个主要原因。首先,Cufflinks和rQuant不能完全处理映射到多个基因的读取(“基因多重读取”)。Cufflinks使用了一种类似于“拯救”的策略来实现多读的初始部分分配,这大致相当于RSEM和IsoEM使用的EM算法的一次迭代。至于rQuant,还不清楚[2这种方法是否以及如何处理基因多重读取。造成性能差距的第二个原因是Cufflinks和rQuant需要对基因组的读取,而不是对转录集的读取。正如我们在实现部分所讨论的,RNA- seq reads与基因组序列的对齐对于真核生物物种是具有挑战性的,它们的RNA转录本是剪接和聚腺苷酸化的。
在RefSeq和ensemble集合上,这些方法的相对性能是相似的,尽管Cufflinks在ensemble集合上的准确性惊人地低。对袖扣结果的进一步研究表明,这种方法对转录本的一个子集产生了异常高的丰度估计。这个子集由比平均片段长度(280个碱基)更短(不包括poly(A) tail)的转录本组成,这表明Cufflinks的当前实现不能正确处理短转录本。
对于PE数据,RSEM和IsoEM具有可比性,但对于SE数据,RSEM略准确。相对于IsoEM而言,RSEM的这种相对较小的改进可能是由于多聚(a)尾处理的更详细实现,这在IsoEM的原始版本中并不存在,直到最近才引入到其软件中。RSEM的当前版本相对于RSEM v0.6的改进是由于SE数据的片段长度建模,这最初由[4以提高准确性。
MAQC数据
在真实数据上对RNA-Seq量化方法进行基准测试是具有挑战性的,因为我们很少知道样本中“真实的”转录丰度。目前,qRT-PCR似乎是产生“金标准”丰度测量的最流行的技术,尽管如果没有仔细的实验设计和数据分析,它可能会给出不准确的结果[35].虽然RNA-Seq被普遍认为是一种比微阵列更精确的量化技术[1],是否也优于qRT-PCR还有待观察。
在我们的测试中,我们使用了微阵列质量控制(MAQC)项目中使用的样本生成的数据[36],正如在其他一些RNA-Seq定量准确性研究中所做的那样[37,38].MAQC项目评估了多种微阵列平台和技术,包括TaqMan qRT-PCR,对两种人类RNA样本进行了评估,其中一种来自脑组织(HBR),另一种来自混合组织类型(UHR)。该项目的TaqMan qRT-PCR测量结果包含了一小部分(1000个)基因的丰度值,对两个样本分别进行了4次技术重复。最近,有三个小组在两个MAQC样本上生成了RNA-Seq数据[25,37,39].
我们将量化方法应用于每个MAQC RNA-Seq数据集,并将其丰度预测与qRT-PCR值进行比较。所有方法都提供了人类RefSeq基因注释。对于模拟实验,Cufflinks只在量化模式下运行,TopHat只允许映射到注释中存在的拼接结。Cufflinks和IsoEM分别使用和不使用它们的序列特定偏差校正模式,这可以提高用随机六聚体启动协议生成的RNA-Seq库的量化精度,该协议用于所有MAQC RNA-Seq数据。我们没有运行带有位置特定偏差校正(RSPD)的RSEM,因为这只适用于oligo-dT引物RNA-Seq文库,这些文库通常偏向于来自转录本3'端的读取。
为了评估RNA-Seq丰度预测与qRT-PCR测量结果的相似性,我们计算了丰度值对数的Pearson相关性。我们使用对数转换来防止相关值被最丰富的转录本所支配。为了避免零的问题,我们只计算那些通过qRT-PCR和所有方法预测丰度非零的基因的相关值。我们还计算了每种方法的假阳性(FP)、真阳性(TP)、假阴性(FN)和真阴性(TN)计数,其中“阳性”表示预测丰度非零,真实性由qRT-PCR测定结果确定。
每一个MAQC RNA-Seq样本上测试方法的相关值如表所示2.一般来说,这些方法为每个样本提供了可比较的相关值。确认[38], Cufflinks的偏差修正模式给出的预测相关性高于其他方法,尤其是在HBR样本上。与Cufflinks不同的是,IsoEM的偏差校正模式对其与这些样品的qRT-PCR值的相关性没有显著影响。在没有对数转换丰度的情况下计算的斯皮尔曼和皮尔森相关值得出了类似的结果3.).这些方法的TP、FP、TN和FN计数也可进行比较(附加文件3.).
这些数据集上的方法(启用偏差校正的袖扣除外)之间缺乏明确的区别,这可以用许多因素来解释。首先,qRT-PCR测量结果仅适用于RefSeq集合中19,005个基因中的1,000个(5%)。在筛选与RefSeq注释一致且丰度非零的qRT-PCR基因(见方法)后,只有716个基因可用于相关分析。第二,这组基因偏向于单亚型基因和序列相对独特的基因,降低了这些数据区分那些更擅长于亚型量化或多读处理的方法的能力。在这组基因中,每个基因的同工型的平均数为1.1,而所有基因的同工型平均数为1.7 (p< 10−115,情绪中位数检验)。类似地,集合中基因的平均“可映射性”(参见方法)为0.96,而所有基因的平均“可映射性”为0.91 (p< 10−6).最后,qRT-PCR值的偏差,可能是由于扩增效率的变化[35],可能导致了金本位的不准确。
运行时间和内存
除了比较量化方法的准确性,我们还测量了它们的运行时间和内存使用量。为此,我们使用了包含2000万个片段的模拟小鼠RefSeq数据集,其大小与Illumina基因组分析仪IIx的单个lane产生的数据相当。表格3.列出了每种方法在SE和PE数据上的运行时间和峰值内存使用量。额外的文件4给出了模拟小鼠ensemble数据集的对应值。所有方法都运行在8核2.93 GHz Linux服务器上,内存为32 GB,并启用了超线程。Bowtie比对转录本序列集和RSEM量化使用的内存最少,大约为1.1 GB。Cufflinks和rQuant的内存使用峰值是由于运行TopHat将读取对齐到基因组。这两种方法的量化程序在RefSeq数据集上分别需要0.4和1.6 GB的内存。IsoEM是最快的方法,但有最大的内存需求,高达14 GB。应该注意的是,这些方法的运行时间不是完全可比较的,因为RSEM和Cufflinks除了计算ML估计之外还计算ci,而其他方法只计算ML估计。
RSEM所需的运行时间和内存与读取对齐的数量成线性关系,而读取对齐的数量通常与读取的数量成正比。虽然当前版本的RSEM有一个并行EM算法,但由于两个原因,它并不比原始版本快。首先,当前版本运行EM算法多次迭代,以提高精度。在这个数据集中,当前版本运行了4802次迭代,而较老的版本运行了643次迭代。其次,当前版本的运行时间包括计算95%可信区间的时间,这需要大量的计算,而不是原始版本的功能。
实验结果
随着RSEM扩展到PE数据模型和带有质量评分信息的读取,我们开始确定这些更复杂的数据类型是否允许提高丰度估计精度。为此,我们进行了两组模拟实验。在第一组实验中,我们比较了PE读取和SE读取的性能。对于第二种方法,我们测试了质量分数是否提供了提高估计准确性的信息。
配对与单端读取
我们先前表明,对于SE RNA-Seq协议,读取的数量比读取的长度更重要,以提高基因水平丰度估计的准确性[7].在固定的测序吞吐量(以碱基总数为标准)下,我们发现在小鼠和玉米中SE RNA-Seq分析的最佳读取长度约为25个碱基。这一结果被后来的一项研究证实了[4].最近的研究得出结论,PE reads比SE reads具有更高的估计精度,特别是对于选择性拼接基因的异构体[3.,4].随着RSEM现在扩展到PE数据模型,我们决定用我们自己的模拟来测试这些结果。
我们用四种不同的配置模拟RNA-Seq数据:(1)2000万,35个碱基SE读取,(2)2000万,70个碱基SE读取,(3)2000万,35个碱基PE读取,(4)4000万,35个碱基SE读取。就测序的碱基数量而言,后三种配置给出了相同的吞吐量,因此在成本方面是最具有可比性的,在一个简单的经济模型中,人们为每个测序的碱基付费。我们对人和小鼠进行了模拟,并使用了RefSeq和ensemble注释,以确定物种或注释集是否是一个因素。除了模拟每种配置的不同物种和注释集外,我们还模拟了有和没有测序错误的情况,以评估变量读对齐灵敏度是否有影响。
表格4给出从RefSeq模拟数据集计算的RSEM估计值的MPE、10% EF和FP(附加文件5给出了ensemble集合的对应值)。正如预期的那样,在读取数量固定的情况下,70个基本读取比35个基本读取提供了更好的估计精度。证实先前的结果[3.,4],在读取数量和总吞吐量固定的情况下,PE读取比SE读取提高了估计精度(将PE精度与SE 70基本精度进行比较)。然而,在相同的测序通量下,短SE reads在基因水平上提供了最高的估计精度。这一结果在两种物种中都成立,无论读取是否包含测序错误。这些结果表明,如果主要目标是准确估计基因丰度,那么对大量短的SE reads进行测序是最好的。例如,如果在一个PE 35碱基读取的Illumina序列和两个SE 35碱基读取的Illumina序列之间进行选择,我们的模拟表明,后者将为基因水平估计提供最佳的总体量化结果。在此场景中使用SE读取的另一个优点是SE读取的两个车道可以并行运行,而PE车道的两端目前是一个接一个生成的。因此,使用短的SE读取可以节省测序时间。这个结果依赖于SE估计过程中提供的片段长度分布,因为SE数据不容易被用来自动确定这个分布。然而,这种分布通常可以通过其他方法提前获得。
另一方面,如果主要的兴趣是在单个基因中替代剪接事件的相对频率,那么PE数据可以根据转录集提供更准确的估计。对于人类RefSeq模拟,PE数据比SE数据显示了更大的准确性改进,这是由人类RefSeq注释平均每个基因(1.6)比小鼠RefSeq注释(1.2)有更多的异构体这一事实解释的。使用ensemble注释的模拟结果进一步支持了这一点,平均每个基因有更多的异型(人类6.3,小鼠3.4)。因此,对于基因经历了大量可选剪接事件的物种,PE数据可能更适合推断这些事件的相对频率。虽然基因水平和基因内异构体水平估计的结果是明确的,但全球异构体水平估计的结果是混合的。在某些模拟集中,SE数据的性能优于PE数据(具有相同的吞吐量),而在其他模拟集中,情况则相反。这可以用这样一个事实来解释,即亚型的整体丰度是其基因丰度和基因内丰度的产物。因此,可以通过在其他两个水平上做出更好的丰度估计来提高全局亚型丰度的准确性。通过更准确的基因水平估计SE数据和通过更准确的基因内亚型估计PE数据,可以改进全局亚型水平估计。
总之,我们建议研究人员在决定测序参数(如读取长度和读取次数)之前仔细考虑他们的RNA-Seq实验的目标。虽然人们可能倾向于产生长序列和PE序列,但如果唯一的目标是量化基因丰度,那么使用大量的SE序列可能更具有成本效益。如果目标是分析基因内异构体频率或执行非量化任务,如转录组组装,则PE读取应优先考虑。为了确定用特定转录本集进行量化的最佳测序策略,可以使用RSEM模拟工具。
RNA-Seq量化的质量分数值
我们进行了模拟实验,以确定是否使用质量评分(而不仅仅是读取序列本身)提高了RNA-Seq数据量化的准确性。进行了两次SE模拟,每一次都采用不同的测序误差模型。模拟使用鼠标RefSeq转录集作为参考。在第一个模拟中,根据给定位置的质量评分的理论误差概率,在给定的读取位置引入一个误差。即在具有Phred质量评分的位置引入错误的概率问是10−问/ 10.在第二个模拟中,给出质量评分的排序错误的概率问是由训练数据确定的(我们称之为“经验”模型)。对于这两个模拟数据集,我们使用两种不同的模型用RSEM估计丰度:一种考虑质量分数(“质量分数”模型),另一种使用我们的原始误差模型,该模型不考虑质量分数,而是估计一个位置和基础相关的测序误差模型(“剖面”模型)。计算了两个RSEM模型在两个模拟数据集上的丰度估计值的MPE、10% EF和FP统计量5).我们发现,即使测序错误遵循由质量分数给出的理论概率,质量分数模型的准确性与剖面模型几乎没有区别。使用ensemble抄本集进行的模拟得到了类似的结果(附加文件6).这表明,为了从RNA-Seq数据进行量化,从illumina生成的读取的质量评分提供的额外信息很少。然而,这并不意味着不需要对测序错误进行建模。相反,这些结果表明,一个有效的测序错误模型可以从读取序列单独学习。我们强调,这些结果仅用于量化。诸如SNP检测等应用程序肯定需要考虑质量评分信息。
结论
我们提出了RSEM,一个从RNA-Seq数据执行基因和亚型水平量化的软件包。通过对真实数据的模拟和评估,我们证明了RSEM比其他量化方法具有更好的性能或相当的性能。与其他工具不同的是,RSEM不需要参考基因组,因此对于从新转录组集合的量化应该是有用的。该软件包对RNA-Seq研究人员还有许多其他有用的功能,包括可视化输出和CI估计。此外,该软件用户友好,通常最多需要两个命令来估计从原始RNA-Seq读取丰度,并使用标准格式的参考转录文件。最后,RSEM的模拟模块对确定定量实验的最佳排序策略是有价值的。利用这个模块的优势,我们已经确定大量的短SE读取是基因水平量化的最佳选择,而PE读取可能提高小鼠和人类转录组的基因内异构体频率。
RSEM将继续发展,以保持最新的测序技术和RNA-Seq协议细节的研究。未来的工作将包括在模型中加入额外的偏差,如特定于序列的读取位置偏好[38,40和转录本特定的读分布[41].我们还打算在读取对齐中添加对ABI SOLiD测序器和索引生成的颜色空间读取的支持。
可用性和需求
项目名称:RSEM
操作系统:任何posix兼容平台(例如,Linux, Mac OS X, Cygwin)
编程语言:c++, Perl
其他要求:Pthreads;领结(29的默认对齐方式为rsem-calculate-expression;R为rsem-plot-model.
许可:GNU GPL。
方法
统计模型
RSEM所使用的统计模型可以用如图所示的有向图形模型表示4.与我们最初的统计模型相比[7,这个模型在四个方面得到了扩展。首先,PE读取现在被建模,使用一对观察到的随机变量,R1而且R2.对于SE的例子,R2作为一个潜在的随机变量。其次,从一个或一对读取派生的片段长度现在被建模,并由潜在随机变量表示F.的分布F是否使用全局片段长度分布指定λF,如果一个片段来自有限长度的特定转录本,它将被截断并规范化。也就是说,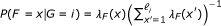 在哪里ℓ我成绩单的长度我.使用片段长度分布进行RNA-Seq定量是由[6]用于对端数据,其后用[4]用于单端数据。
在哪里ℓ我成绩单的长度我.使用片段长度分布进行RNA-Seq定量是由[6]用于对端数据,其后用[4]用于单端数据。

RSEM使用的有向图形模型.该模型包括N随机变量集,每个测序的RNA-Seq片段一个。的片段n,其父转录本、长度、起始位置和方向用潜变量表示Gn,Fn,年代n而且On分别。对于PE数据,观察到的变量(阴影圈)为读取长度(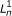 而且
而且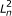 )、品质评分(
)、品质评分(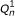 而且
而且 )和序列(
)和序列(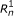 而且
而且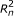 ).对于SE数据,,,未被注意的。模型的主要参数由向量给出θ,表示从每个转录本派生出的片段的先验概率。
).对于SE数据,,,未被注意的。模型的主要参数由向量给出θ,表示从每个转录本派生出的片段的先验概率。
第三个扩展允许不同的读取长度(例如454数据)。读取的长度由观察到的随机变量表示l(或l1而且l2为体育阅读)。与片段长度模型相似,的分布l是否使用全局读长度分布指定λR,在给定特定片段长度的情况下截断并规范化。在符号,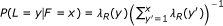 .最后,读取的质量分数现在被用来模拟该读取序列的概率。读取的质量评分字符串由随机变量表示问.为量化的目的,我们不指定的分布问随机变量,因为它们被观察到,不依赖于任何其他随机变量(也就是说,我们只对给定质量分数的读取的条件似然感兴趣)。我们不依赖质量分数隐含的理论误差概率,而是使用经验误差函数,ε.给定读取位置我有质量评分问我由参考字符派生而来c,读取字符的条件概率r我是P(r我|问我c) =ε(r我,问我c).如果质量分数不可得或不可靠,则我们的位置和参考字符相关误差模型[7可以使用。
.最后,读取的质量分数现在被用来模拟该读取序列的概率。读取的质量评分字符串由随机变量表示问.为量化的目的,我们不指定的分布问随机变量,因为它们被观察到,不依赖于任何其他随机变量(也就是说,我们只对给定质量分数的读取的条件似然感兴趣)。我们不依赖质量分数隐含的理论误差概率,而是使用经验误差函数,ε.给定读取位置我有质量评分问我由参考字符派生而来c,读取字符的条件概率r我是P(r我|问我c) =ε(r我,问我c).如果质量分数不可得或不可靠,则我们的位置和参考字符相关误差模型[7可以使用。
Expectation-Maximation
给定一组RNA-Seq数据,RSEM的主要目标是计算参数的ML值,θ,为上一节给出的模型,其中θ我表示一个片段来自转录本的概率我(与θ0表示“噪声”转录本,从中可以派生没有对齐的读取)。一旦估计,θ值被转换为转录分数(我们用τ)使用方程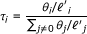 在哪里
在哪里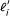 成绩单的有效长度我[6],由
成绩单的有效长度我[6],由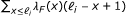 对于poly(A)-转录本和
对于poly(A)-转录本和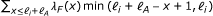 对于poly(A)+转录本,其中ℓ一个为poly(a)尾巴的长度。有效长度可以被认为是一个片段在转录序列中可能开始的位置的平均数我.
对于poly(A)+转录本,其中ℓ一个为poly(a)尾巴的长度。有效长度可以被认为是一个片段在转录序列中可能开始的位置的平均数我.
RSEM计算近似的ML估计θ使用EM算法(详情请参阅[7])。这些估计是近似的,因为使用对齐来限制可能的位置,从这些位置可以得到读取。在EM的前20次迭代(以及每100次迭代)中,片段长度、RSPD和测序误差分布的参数随着θ.在所有其他迭代过程中,只有θ完成参数更新。这种估计策略是对RSEM最初实现的改进,它估计除θ在EM之前使用唯一映射读取。当所有θ我值≥10−7相对变化小于10−3.收敛后,RSEM输出MLτ值,以及在给定ML参数的情况下,从每个转录本派生的RNA-Seq片段数量的期望值。
为了加快推断速度,将过滤大量对齐的读取(默认情况下至少200)。此外,我们还过滤掉了可能来自poly(A)尾的读数,因为对齐器可能并不总是检测到这些读数有很多对齐。由于对齐近似和这种过滤策略,所述EM过程的直接应用将导致对包含高度重复序列(包括聚(a)尾)的转录本的偏丰度估计。因此,我们对ML估计器稍加修改,以调整这个偏差。的成绩单我,我们计算一个值米我,即从抄本生成读(片段)的概率我不会有大量的对齐。一般来说,的值米我取决于片段长度分布、读取长度分布、RSPD、协议的链特异性和聚(a)尾的长度。在EM的最大化步骤中,我们的修改是设置θ我正比于c我/(纳米我),c我预期的片段数是否来自转录本我而且N是未过滤片段的总数。
吉布斯抽样
除了计算ML估计外,RSEM还使用其模型的贝叶斯版本来计算PME和丰度的95% ci。在贝叶斯模型中θ参数被视为具有狄利克雷先验分布的潜在随机变量。Dirichlet分布的参数(α)设为1,这使得先验等价于均匀分布,最大的后验估计θ等于ML估计值。
RSEM采用两阶段采样过程计算pme和95% ci。首先,折叠吉布斯采样算法的一个标准应用[42]用于获取计数向量的采样集,其中每个向量表示映射到每个转录本的片段的数量。在每一轮吉布斯采样算法中,每个片段的真实映射将根据所有其他片段的当前映射重新采样。根据EM算法计算的ML参数对每个片段的初始映射进行采样。该算法对1000个计数向量进行采样。
抽样过程的第二阶段涉及到的抽样值θ给定从第一阶段采样的每个计数向量。给定一个计数向量,c,一个θ向量从它的后验分布中采样,它是一个简单的狄利克雷分布α我=c我+ 1。对于每个计数向量,50θ对载体进行采样,得到50000个样本θ.的θ样本转换为转录分数(τ),然后进行总结,得出每个转录本丰度的PME和95% CI。
为了验证RSEM生成的ci,我们模拟了一个带有鼠标RefSeq注释的RNA-Seq数据集,并估计了RSEM生成的ci的可信度从50%到95%。然后,我们计算了在所有丰度至少为1 TPM的转录本中,真实丰度落在可信区间内的转录本的比例(表6).结果表明,95%可信区间是相当准确的,而且这些区间很紧(因为正确预测的转录水平的比例随着可信水平的下降而下降)。用鼠标ensemble注释模拟的数据估计的ci不太准确(附加文件7).我们调查了为什么ci在这个集合上不那么准确,发现由于狄利克雷先验和在ensemble集合中大量的转录本,许多ci是向下偏斜的。尽管对于ensemble集合的ci没有那些对于RefSeq集合的表现那么好,我们预计它们在比较样本间的丰度时仍然有用,因为ci中的偏差应该是一致的。然而,这些结果表明,需要进一步的工作来开发先验分布,以更好地处理RNA-Seq数据集中典型的大量零丰度转录本。
参考序列
参考转录本集注释使用了两个来源:来自UCSC基因组浏览器数据库的RefSeq基因注释[26和发布63个注解[27].用于人类和小鼠RefSeq注释的基因组版本分别为build 36.1 (UCSC hg18)和build 37 (UCSC mm9)。对于ensemble bl人类注释,使用build 37 (UCSC hg19)代替。对RefSeq和Ensembl注释进行过滤,去除非编码基因和位于非标准染色体上的基因(如chr1_random和chr5_h2_hap1)。此外,我们还鉴定了一小部分位于多个不重叠位置的RefSeq基因,并将它们重新命名,以便每个基因起源于一个独特的位点。
模拟
RSEM使用的生成统计模型很容易用于模拟RNA-Seq数据。除了模型的主要参数(如丰度、片段和读取长度分布以及测序误差模型参数),还必须提供质量评分信息以模拟读取。对于本文的模拟目的,我们使用质量分数的一阶马尔可夫链模型为每次阅读生成质量分数字符串。模拟模型的参数来源于序列读取档案(Sequence Read Archive, SRA)中的真实RNA-Seq数据集。小鼠模拟参数从SRA登录SRX026632中获得,其中包含约420万个PE 35碱基reads,测序自C2C12小鼠成肌细胞的poly(a)+ RNA文库[3.].对于人体模拟,我们从SRA接入SRX016368中学习了参数,其中包括从MAQC UHR样本中测序的约9300万个SE 35碱基reads [37].由于人类数据是SE读取,RSEM提供了一个片段长度分布μ= 200和σ= 29,以便学习其他模型参数。然而,在模拟中,人类和老鼠的数据都是用片段长度分布生成的μ= 280和σ= 17,用于[3.]进行类似的模拟。最后,为了模拟mrna具有poly(A)尾这一事实,我们在每个转录本的末尾添加了125个A。
MAQC验证
TaqMan qRT-PCR检测结果下载于基因表达综合(GEO)(平台GPL4097)。对于每个样本,一个基因的丰度被作为所有技术复制中分配给该基因的所有探针通过检测阈值的平均值。后(37],如果75%的探针通过检测阈值,则认为该基因已表达。GEO记录中列出的每个基因的RefSeq转录本接入与基因组注释中每个基因的RefSeq接入进行比较。只有GEO位点是注释位点超集的基因被保留。这样做是为了确保RNA-Seq估计值与qRT-PCR探针的值相比较,而qRT-PCR探针只能保证与GEO记录中给出的接入相对应。筛选结果为716个基因,其中在UHR和HBR中分别检测到656和618个基因。
为了分析筛选的qRT-PCR基因对整个人类RefSeq基因集的代表性,我们计算了每个基因的“可映射性”。对于每个亚型,我们从其序列中生成所有可能的35个碱基读取,并用Bowtie将它们与整个转录本集合对齐,最多允许两次不匹配。同源异构体的可映射性计算为仅与其基因的同源异构体相匹配的reads的比例。基因的可映射性被计算为其亚型可映射性的平均值。
缩写
- 体育:
-
paired-end
- SE:
-
单头
- ML:
-
最大似然
- 中外职业:
-
后验均值估计
- 置信区间:
-
可信度区间
- 迈普:
-
中位数百分比误差
- 英孚:
-
错误的部分
- 外交政策:
-
假阳性
- RSPD:
-
读取起始位置分布
参考文献
王铮,Gerstein M, Snyder M: RNA-Seq:转录组学的革命性工具。自然评论,2009,10:57-63。10.1038 / nrg2484
Bohnert R, Rätsch G: rQuant。基于rna - seq的转录本定量的工具。核酸研究2010,(38 Web Server): W348-51。
卡兹Y,王等,Airoldi EM, Burge CB:用于识别亚型调控的RNA测序实验分析与设计。自然方法,2010,7(12):1009-15。10.1038 / nmeth.1528
Nicolae M, Mangul S, mannidoiu I, Zelikovsky A:从RNA-Seq数据中估计可选剪接异构体频率。生物信息学中的算法,计算机科学课堂讲稿。编辑:莫尔顿V,辛格M.利物浦,英国:施普林格柏林/海德堡;2010:202 - 214。
蒋宏,王文华:RNA-Seq中异构体表达的统计推断。生物信息学,2009,25(8):1026-1032。10.1093 /生物信息学/ btp113
Trapnell C, Williams B, Pertea G, Mortazavi A, Kwan G, van Baren M, Salzberg S, Wold B, Pachter L: RNA-Seq对转录本的组装和量化揭示了细胞分化过程中未注释的转录本和亚型转换。自然生物技术,2010,28(5):511-515。10.1038 / nbt.1621
Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN:具有读映射不确定性的RNA-Seq基因表达估计。生物信息学,2010,26(4):493-500。10.1093 /生物信息学/ btp692
序列计数数据的差分表达式分析。基因组生物学2010,11(10):R106..
Robinson MD, McCarthy DJ, Smyth GK: edgeR:用于数字基因表达数据的差异表达分析的Bioconductor包。生物信息学,2010,26:139-40。10.1093 /生物信息学/ btp616
Guttman M、Garber M、Levin JZ、Donaghey J、Robinson J、Adiconis X、Fan L、Koziol MJ、Gnirke A、Nusbaum C、Rinn JL、Lander ES、Regev A:小鼠细胞类型特异性转录组从头计算重建揭示了lincRNAs保守的多外胞子结构。自然生物技术,2010,28(5):503-510。10.1038 / nbt.1633
Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, Mungall K, Lee S, Okada HM, Qian JQ, Griffith M, Raymond A, Thiessen N, Cezard T, Butterfield YS, Newsome R, Chan SK, She R, Varhol R, Kamoh B, Prabhu AL, Tam A, Zhao Y, Moore RA, Hirst M, Marra MA, Jones SJM, Hoodless PA, Birol I: RNA-seq数据的重新汇编和分析。自然方法2010,7(11):909-12。10.1038 / nmeth.1517
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson Da, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, haacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, lindbladal - toh K, Friedman N, Regev A:从没有参考基因组的RNA-Seq数据中组装完整的转录组。自然生物技术,2011,29(7):644-52。10.1038 / nbt.1883
王卓,王志强,王文华,王志强,王志强,王志强,王志强,王志强,王志强,王志强,王志强,王志强,王志强,王志强:酵母基因组转录序列分析。科学通报,2008,32(4):344 - 349。10.1126 / science.1158441
Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y: RNA-seq:技术重现性评估和与基因表达阵列的比较。基因组学研究2008,18(9):1509-17。10.1101 / gr.079558.108
Morin R, Bainbridge M, Fejes A, Hirst M, Krzywinski M, Pugh T, McDonald H, Varhol R, Jones S, Marra M:使用随机引物cDNA和大规模并行短读测序分析HeLa S3转录组。生物技术2008,45:81-94。10.2144 / 000112900
王鑫,吴铮,张鑫:亚型丰度推断为RNA-seq中基因表达水平的估计提供了更精确的方法。生物信息学与计算生物学2010,8(增刊1):177-92。
Faulkner GJ, Forrest ARR, Chalk AM, Schroder K, Hayashizaki Y, Carninci P, Hume DA, Grimmond SM:对multimapping短序列标签的拯救策略改进了CAGE对转录活性的调查。基因组学研究,2008,29(3):381 - 381。10.1016 / j.ygeno.2007.11.003
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B:利用RNA-Seq绘制和量化哺乳动物转录组。自然方法2008,5(7):621-8。10.1038 / nmeth.1226
冯军,李伟,姜涛:从短序列reads推断异构体。计算生物学报,2011,18(3):305-21。10.1089 / cmb.2010.0243
payananiuc B, Zaitlen N, Halperin E: RNA-seq实验中同源基因表达水平的准确估计。计算生物学报,2011,18(3):459-68。10.1089 / cmb.2010.0259
Richard H, Schulz MH, Sultan M, Nürnberger A, Schrinner S, Balzereit D, Dagand E, Rasche A, Lehrach H, Vingron M, Haas SA, Yaspo ML:从RNA-Seq实验中的外显子表达水平预测替代异构体。核酸研究,2010,38(10):e112。
Taub M, Lipson D, Speed TP:分配歧义短读的方法。通信与信息系统,2010,10(2):69-82。
De Bona F, Ossowski S, Schneeberger K, Ratsch G:短序列读取的最佳拼接对齐。生物信息学2008,24 (16):i174-180。10.1093 /生物信息学/ btn300
Trapnell C, Pachter L, Salzberg SL: TopHat:发现RNA-Seq剪接。生物信息学,2009,25(9):1105-11。10.1093 /生物信息学/ btp120
Au KF,蒋宏,林玲,邢颖,王文华:基于SpliceMap的RNA-seq配对数据剪接连接检测。核酸研究,2010,38(14):4570-8。10.1093 / nar / gkq211
Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, Diekhans M, Dreszer TR, Giardine BM, Harte RA, Hillman-Jackson J, Hsu F, Kirkup V, Kuhn RM, Learned K, Li CH, Meyer LR, Pohl A, Raney BJ, Rosenbloom KR, Smith KE, Haussler D, Kent WJ: UCSC基因组浏览器数据库:2011更新。核酸研究2011,(39数据库):D876-82。
Flicek P, Amode先生,巴雷尔D,比尔K,布伦特年代,陈Y,克拉珀姆P,科茨克,Fairley年代,菲茨杰拉德,戈登L,亨德里克斯米,每小时T,约翰逊N, Kahari, Keefe D,基南年代,Kinsella R, Kokocinski F, Kulesha E,拉尔森P, Longden我,麦克拉伦W, Overduin B, Pritchard B, Riat HS,里奥斯D,里奇GRS, ruffy M,舒斯特尔M, Sobral D, Spudich G,唐丫,Trevanion年代,Vandrovcova J, Vilella AJ,白色的年代,怀尔德SP, Zadissa,扎莫拉J, Aken提单,伯尼E, F坎宁安,邓纳姆,杜宾R,Fernández-Suarez XM, Herrero J, Hubbard TJP, Parker A, Proctor G, Vogel J, Searle SMJ: ensemble 2011。核酸研究2011,(39数据库):D800-6。
Roberts A, Pimentel H, Trapnell C, Pachter L:利用RNA-Seq识别注释基因组中的新转录本。2011年生物信息学。2011年6月21日首次在线出版
Langmead B, Trapnell C, Pop M, Salzberg SL:人类基因组短DNA序列的超快和高效记忆对齐。基因组生物学2009,10(3):R25..
李H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R:序列对齐/地图格式和SAMtools。生物信息学,2009,25(16):2078-9。10.1093 /生物信息学/ btp352
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler, David: UCSC的人类基因组浏览器。基因组学研究,2002,12(6):996-1006。
李娟,蒋宏,王文华:RNA-Seq数据中短读速率的非均匀性建模。基因组生物学2010,11(5):R50..
Pruitt KD, Tatusova T, Klimke W, Maglott DR: NCBI参考序列:当前状态,政策和新举措。核酸研究2009,(37数据库):D32-6。
Bustin SA:为什么需要qPCR出版指南?- MIQE的案例。方法学报2010,50(4):217-26。10.1016 / j.ymeth.2009.12.006
施L,里德LH,琼斯WD, Shippy R,沃灵顿是的,贝克SC,科林斯PJ, de Longueville F,川崎,李肯塔基州,罗Y,太阳丫,Willey JC, Setterquist RA,费舍尔通用、通W,德拉甘YP,迪克斯DJ, Frueh弗兰克-威廉姆斯,Goodsaid调频,赫尔曼·D,詹森房车,约翰逊CD, Lobenhofer EK,宫殿RK, Schrf U, Thierry-Mieg J,王C,威尔逊M, Wolber PK,张L,黑龙江,保W, Barbacioru CC,卢卡斯AB Bertholet V,两C,布罗姆利B, D,布朗Brunner,卡纳莱斯R,曹XM, Cebula助教,陈JJ,程J,楚TM, Chudin E,科森J,葡萄酒JC, cron LJ,戴维斯C,戴维森TS, Delenstarr G,邓X, Dorris D,雷欧AC, Xh粉丝,方H, Fulmer-Smentek年代,Fuscoe JC,加拉格尔K,通用电气W,郭,郭X,海格J, Haje PK,汉族,汉族T, Harbottle HC,哈里斯SC, Hatchwell E,豪泽CA,海丝特年代,香港H, Hurban P,杰克逊SA,霁H,骑士CR、郭WP,勒克莱尔我,利维年代,李求刘C,刘Y, Lombardi MJ,马Y, Magnuson SR, Maqsodi B, McDaniel T,梅N, Myklebost啊,宁B, Novoradovskaya N,奥尔女士,奥斯本TW, Papallo,Patterson T:微阵列质量控制(MAQC)项目显示了基因表达测量的平台间和平台内可重复性。生物技术学报,2006,24(9):1151-61。10.1038 / nbt1239
Bullard JH, Purdom E, Hansen KD, Dudoit S:评估mRNA-Seq实验中归一化和差异表达的统计方法。生物信息学报2010,11:94。10.1186 / 1471-2105-11-94
Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L:通过修正片段偏差提高RNA-Seq表达估计。基因组生物学2011,12(3):R22..
Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB:人类组织转录组中的替代亚型调控。自然科学,2008,456(7221):470-6。10.1038 / nature07509
Hansen KD, Brenner SE, Dudoit S:随机六聚体启动引起的Illumina转录组测序偏差。核酸研究,2010,38(12):e131。
吴铮,王鑫,张鑫:利用非均匀读分布模型改进RNA-Seq中异构体表达推断。生物信息学,2011,27(4):502-8。10.1093 /生物信息学/ btq696
刘建军:贝叶斯计算中的折叠吉布斯采样器及其在基因调控问题中的应用。美国统计学会学报1994,89(427):958-966。10.2307 / 2290921
鸣谢及资助
我们感谢Victor Ruotti、Ron Stewart、Angela Elwell和Jennifer Bolin对该软件的反馈意见和关于RNA-Seq协议的宝贵讨论。我们也感谢该手稿的审稿人所提出的建设性意见。BL由James Thomson博士的麦克阿瑟教授奖和Morgridge生物与医学计算与信息学研究所提供部分资助。CD部分由NIH拨款1R01HG005232-01A1支持。
作者信息
作者和隶属关系
相应的作者
额外的信息
作者的贡献
BL编写了RSEM软件,共同开发了方法学和实验,进行了计算实验,并帮助起草了手稿。CD共同开发了方法论和实验,并撰写了手稿。所有作者阅读并批准了最终稿件。
电子补充材料
权利与权限
本文由BioMed Central Ltd.授权发布。这是一篇开放获取文章,根据创作共用授权协议(http://creativecommons.org/licenses/by/2.0),它允许在任何媒体上不受限制地使用、分发和复制,只要原著被恰当地引用。
关于本文
引用本文
Li, B, Dewey, C.N. RSEM:有或没有参考基因组的RNA-Seq数据的准确转录物定量。BMC生物信息学12, 323(2011)。https://doi.org/10.1186/1471-2105-12-323
收到了:
接受:
发表:
DOI:https://doi.org/10.1186/1471-2105-12-323
关键字
- 丰度估计
- 数向量
- 片段长度分布
- 读长度分布
- 成绩单分数