条文本

摘要
目标非酒精性脂肪性肝病(NAFLD)可持续在单纯肝脂肪变性阶段或进展为脂肪性肝炎(NASH),并增加肝硬化和癌症的风险。我们检查了控制发展到严重NASH的机制,以便为这种疾病制定未来的治疗策略。
设计在NAFLD患者的肝脏、培养的和原代肝细胞以及具有不同肝细胞特异性转录因子表达的转基因小鼠中检测NFATc1的激活和调控(Alb-cre, NFATc1一个.而且NFATc1Δ/Δ).分别饲喂高脂西方日粮(WD)或与NAFLD候选药物牛磺酸去氧胆酸(TUDCA)联合喂养。对nfatc1依赖性内质网应激反应、NLRP3炎症小体激活和疾病进展进行体外和体内评估。
结果NFATc1在健康肝脏中表达较弱,但在晚期NAFLD中强烈诱导,它与肝酶值以及肝脏炎症和纤维化相关。此外,高脂肪WD增加了NFATc1的表达、核定位和激活,促进了NAFLD的进展,而肝细胞特异性转录因子的减少可以防止小鼠疾病加速。机制上,NFATc1通过内质网应激感应和激活PERK-CHOP未折叠蛋白反应(UPR)驱动肝细胞损伤和炎症。最后,TUDCA可以阻断nfatc1诱导的疾病向NASH的进展。
结论NFATc1通过慢性内质网应激感知和随后肝细胞末端UPR信号的激活,刺激NAFLD进展。例如,通过TUDCA干预内质网应激反应,可以保护脂肪肝不向明显的NASH发展。
- 非酒精性脂肪肝炎
- 脂肪肝
- 肝纤维化
- 炎症
数据可用性声明
资料应合理要求提供。所有与研究相关的数据都包含在文章中或作为补充信息上传。
这是一篇开放获取的文章,按照创作共用署名非商业性(CC BY-NC 4.0)许可发布,该许可允许其他人以非商业性的方式发布、混编、改编、构建本作品,并以不同的条款授权他们的衍生作品,前提是原创作品被正确引用,给予适当的荣誉,任何更改都被注明,且使用是非商业性的。看到的:http://creativecommons.org/licenses/by-nc/4.0/.
来自Altmetric.com的统计
本研究的意义
关于这个问题,我们已经知道了什么?
非酒精性脂肪肝(NAFLD)是慢性肝病的主要原因。
暴露于脂毒性脂肪酸可引起肝细胞内质网(ER)应激。
慢性未解决的内质网应激驱动NAFLD进展。
新的发现是什么?
NFATc1在晚期NAFLD中高度激活。
脂毒性脂肪酸刺激NFATc1在肝细胞中的表达、核定位和激活。
NFATc1的激活通过慢性内质网应激感应和激活末段PERK-CHOP未折叠蛋白反应(UPR)通路驱动肝细胞损伤和炎症。
牛磺酸去氧胆酸(TUDCA)抑制慢性内质网应激反应阻断NFATc1介导的终末UPR信号,并阻止NAFLD进展。
在可预见的未来,它会对临床实践产生怎样的影响?
我们的研究结果表明NFATc1在慢性内质网应激反应和NAFLD进展中起关键作用。靶向不受限制内质网应激可减轻nfatc1驱动的细胞损伤,因此,我们的研究为目前针对TUDCA治疗NAFLD潜力的临床试验提供了依据。
简介
非酒精性肝病(NAFLD)正在成为慢性肝病的主要原因,估计在全世界的患病率超过25%。1 2NAFLD常与代谢紊乱相关,如胰岛素抵抗、2型糖尿病或肥胖,通常表现为单纯的肝脂肪变性(NAFL)。然而,约20%的患者可进展为非酒精性脂肪性肝炎(NASH),这增加了发展为肝硬化、肝衰竭和肝细胞癌的风险。3 4近年来,人们做出了巨大的努力来开发治疗NAFLD的药物策略,但直到目前为止,还没有批准的药物用于临床使用的NASH的预防或治疗。
缺乏有效的治疗主要是由于对导致疾病发展和进展的分子机制的了解仍然不完全。肝细胞中脂肪酸和脂毒性代谢物的过度积累被认为是NAFLD起始的关键事件。5 - 7例如,已有研究表明,持续暴露于脂毒性脂肪酸可通过诱导慢性内质网(ER)应激和随后激活末端未折叠蛋白反应(UPR)途径引起肝细胞损伤。UPR反过来激活NLRP3多蛋白炎症小体复合体,促进肝细胞死亡和炎症反应,以应对未解决的内质网应激。7 - 10因此,在NASH患者的肝脏活检中发现NLRP3炎症体标记物(NLRP3, Caspase-1, Caspase-11,白细胞介素(IL)-1β和IL-18)的高表达。11日12然而,在脂肪肝中负责内源性内质网应激感知和NLRP3激活的内源性通路仍然很大程度上未知。
活化T细胞核因子(NFAT)蛋白包括钙响应转录因子(NFATc1-NFATc4)家族,参与调节免疫系统内外的适应性细胞功能。12日13NFAT蛋白的激活导致对细胞外刺激的反应和对多种细胞应激信号的适应。此外,在代谢性疾病(如糖尿病)、炎症性疾病(如牛皮癣)和恶性疾病(如胰腺癌和黑色素瘤)中也描述了个别家族成员的异位激活。12的程度当非活性时,NFAT蛋白以高磷酸化状态驻留在细胞质中。随着胞质钙的积累,NFAT蛋白被磷酸酶钙调神经磷酸酶去磷酸化并进入细胞核,在细胞核中它们与其他位点特异性转录因子和染色质重构蛋白协同调节靶基因信号。12最近的一些研究表明,NFAT在慢性肝炎和肝癌中的表达和激活水平较高。例如,已有研究表明,NFATc4促进肝细胞PPARα的表达,并通过激活肝星状细胞刺激肝纤维化。22 - 24
在这里,我们首次描述了NFATc1在NAFLD进展中的因果作用。我们发现NFATc1通过NLRP3炎症小体途径促进慢性内质网应激感应和细胞损伤反应。此外,我们还提供了实验证据,证明药物干扰内质网末梢应激反应可能会阻止nfatc1驱动的NAFLD进展。
材料和方法
人类的样本
从哥廷根大学医学中心病理学研究所的生物库中提取了经福尔马林固定、石蜡包埋的NASH患者人体组织。早期脂肪变性患者的正常肝脏切片作为对照。所有样本进行NFATc1免疫组化分析,使用imageJ软件进行分析。简单地说,每个样本10张图像中NFATc1核阳性肝细胞的百分比(比例尺=50µm)是手工计算的。NAFLD活性评分(NAS)和纤维化程度的测定如前所述。25NFATc1在NASH患者中的表达与NAS、纤维化程度及血清中肝酶(ALT和AST)水平存在简单线性回归相关。
动物模型
所有动物实验均根据欧洲实验动物科学协会联合会(Laves批准No. 33.9-42502-04-16/2189和33.9-42502-04-14/1633)的规定批准并进行。Alb-Cre,NFATc1而且NFATc1Δ/Δ之前已经描述过老鼠。19个代谢途径的c.a.NFATc1敲入菌株是通过克隆一个n端血凝素(HA)标记的NFATc1的组成活性版本,在保守的富含丝氨酸的区域包含丝氨酸到丙氨酸的替换,所有三个丝氨酸-脯氨酸重复序列到ROSA26启动子位点(Artemis Pharmaceuticals)而生成的。这些菌株是杂交产生的Alb-Cre; NFATc1Δ/Δ而且Alb-Cre; NFATc1一个.(以下简称NFATc1Δ/Δ而且NFATc1一个老鼠,分别)。如前所述,所有小鼠均采用PCR进行基因分型。19
RNA-Seq
总RNA从转染ha标记的c.n.NFATc1或对照质粒的AML12细胞中分离。用凝胶电泳验证RNA样品的纯度和完整性。来自Illumina的TruSeq RNA文库准备试剂盒(RS-122-2001和RS-122-2002)根据制造商的说明用于制备文库,测序由德国哥廷根大学医学中心NGS综合基因组学核心单元执行。
获取的FastQ文件使用usegalax .eu进行分析,29分析FastQ文件进行质量控制,然后使用TopHat工具将其映射到小鼠转录组(mm9),30.具有非常敏感的bowtie2设置,其次是HTSeq (V.0.9.1;-f bam -r pos -s reverse -a 10 -t exon -m union)。31用DESeq2 (V.2.11.40.6)进行差异基因表达分析,32以及主成分分析(PCA)。热图在GraphPad prims V.7.0中生成。nfatc1依赖性差异表达基因(log2倍值≥0.5/≤−0.5;p≤0.05;基础平均>10)在reactome (reactome.org)通路数据库中分析。
动物治疗的详细描述和进一步的材料和方法包括在在线补充信息.
结果
NFATc1在进展性人和小鼠NAFLD中的核激活
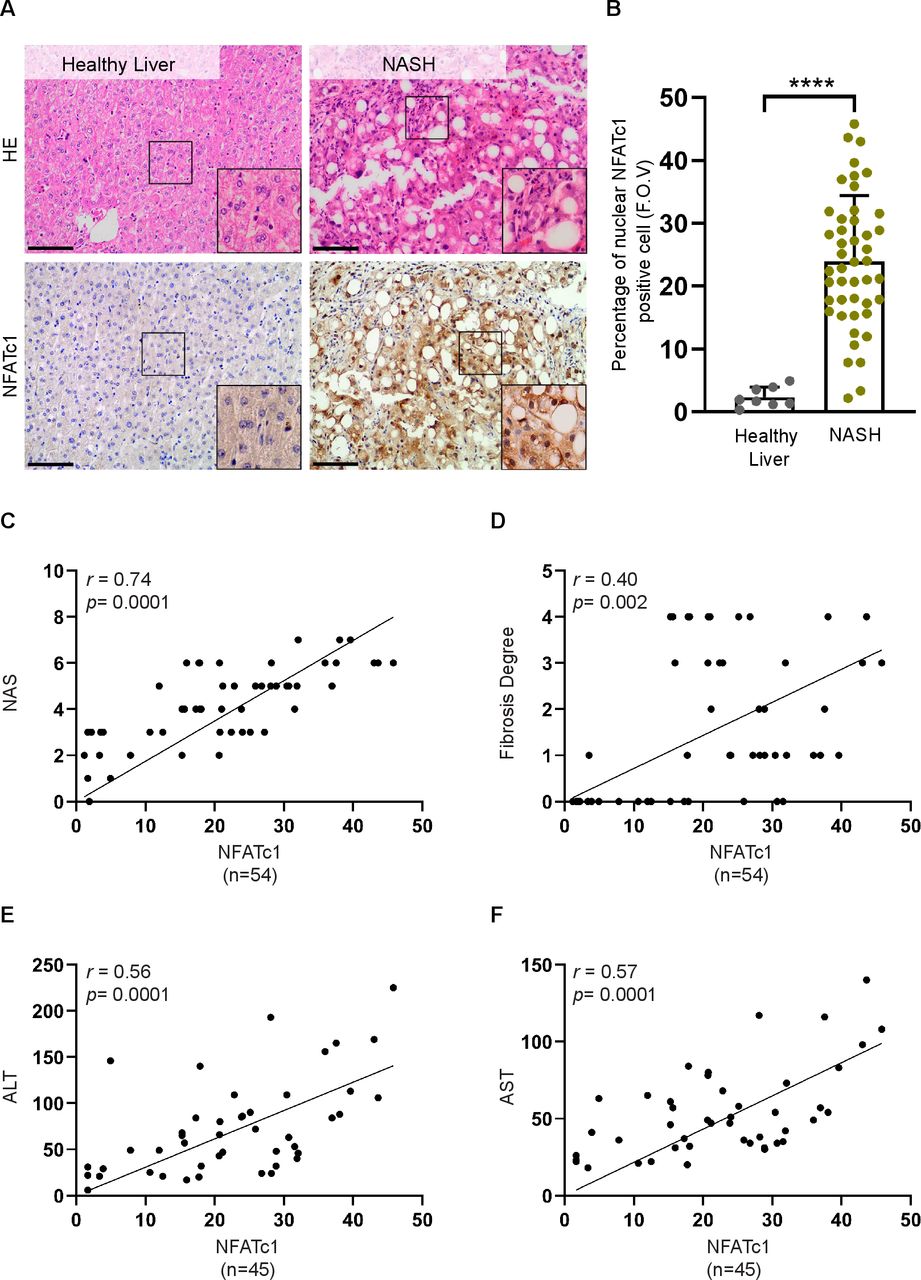
为了评估NFATc1在NAFLD进展中的表达和激活,我们对来自健康肝脏(n=8)和进展性NAFLD (n=46)患者的组织样本进行了免疫组化分析。这些研究表明,在健康肝脏中NFATc1表达缺失或弱,而在进展性疾病患者的肝活检中,NFATc1核表达强烈诱导,其特征为大泡性脂肪变性和明显的小叶炎症(图1 a, B).与NFATc1在疾病加速中的作用一致,我们观察到肝内NFATc1表达与NAFLD进展的显著相关性,由NAS、纤维化程度和NASH患者血液样本中的肝酶水平决定。图1氟).根据这些发现,我们扩大了NFATc1在NAFLD进展中的激活的分析,并研究了NFATc1在进展性NAFLD小鼠模型中的诱导和核定位。为此,我们用高脂肪西方饮食(WD)喂养小鼠20周(图2一个).该模型是研究NAFLD进展渐进步骤的极好工具。33与以前的报道一致,WD治疗的小鼠出现肝脂肪变性,肝细胞呈球囊状,轻度至中度小叶炎症,cd45阳性免疫细胞大量聚集,显著纤维化(图2罪犯).此外,与我们在进展性人类NAFLD中的发现一致,免疫组化和western blot分析证实了NFATc1在NASH肝脏的肝细胞中有强烈的诱导和核积累(图2 e, F).总之,这些在进行性人类和小鼠NAFLD中进行的研究首次提供了NFATc1激活在疾病加速中的作用的实验证据。
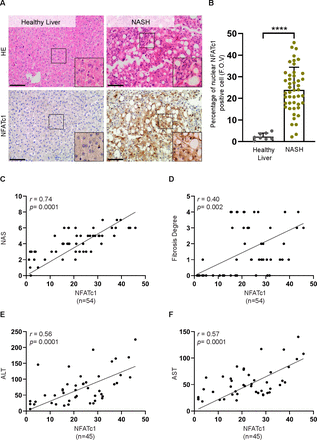
进展性非酒精性肝病(NAFLD)中肝细胞特异性NFATc1的激活(A)健康人肝脏(n=8)和非酒精性脂肪性肝炎(NASH) (n=46)切片用H&E染色和免疫组化方法分析NFATc1。有代表性的图像,比例尺=50µm。(B)健康肝脏和NASH样本中每个视野nfatc1阳性核肝细胞的百分比。统计分析采用非配对t检验。数据以平均值±SD表示,****p≤0.0001。简单线性回归分析显示,肝脏NFATc1表达水平与(a) NAS (NAFLD活性评分)、(B)纤维化程度、(C) ALT水平和(D) AST水平显著相关。

在西方饮食(WD)喂养的小鼠中,NFATc1的诱导与非酒精性脂肪肝(NAFLD)的进展。(A)饲料喂养方案示意图。8周龄C57BL/6野生型小鼠分别给予对照日粮(CD)和WD治疗20周。(B)处死小鼠,肝切片进行H&E染色和免疫组化分析(C) CD45、(D)苦参红和(E) NFATc1的表达。结果具有代表性。比例尺条= 100µm。进行定量分析,结果显示cd45阳性细胞百分比、苦甘草红染色面积百分比和核nfatc1阳性肝细胞百分比来自cd处理和wd处理小鼠的肝脏切片(n=5)。统计分析采用非配对t检验。数据显示在±SD, * * * * * p < 0.005, p < 0.0005, * * * * p < 0.0001。(F)经CD (n=3, lane 1-3)或WD (n=3, lane 4-6)治疗的20周龄小鼠的肝组织裂解物中NFATc1表达的代表性western blot。 Each lane represents liver lysates from individual mice. WD-treated mice express high levels of active NFATc1, indicated by strong increase of the lower band.
NFATc1激活驱动脂肪酸诱导的脂肪性肝炎(NASH)和纤维化
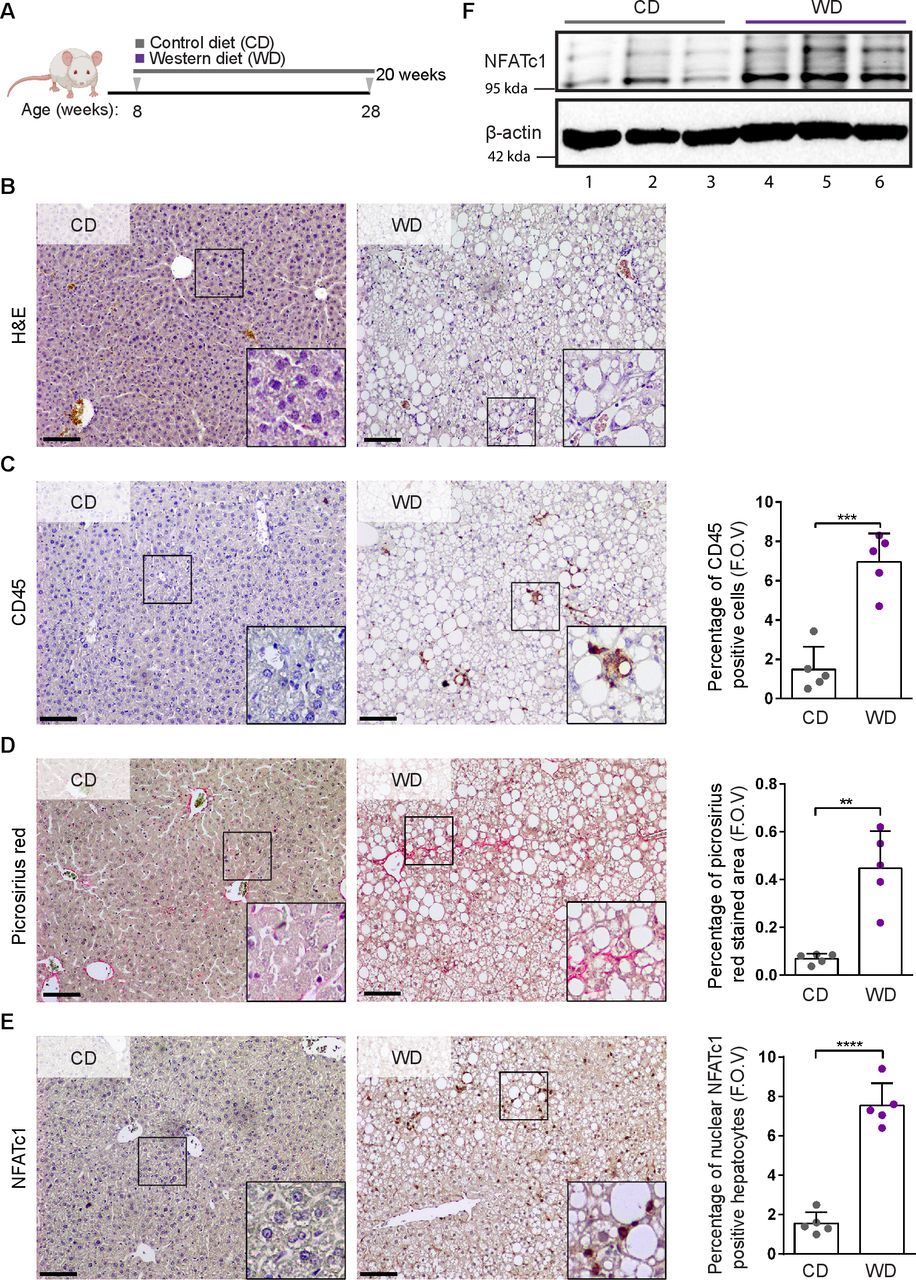
为了研究NFATc1核激活是否确实与NAFLD的起始和进展有因果关系,我们制造了具有肝细胞特异性核激活(Alb-Cre;NFATc1一个.(NFATc1一个.))或耗竭(Alb-Cre;NFATc1Δ/Δ(NFATc1Δ/ΔNFATc1))。内源性NFATc1表达的动物(Alb-Cre)作为对照。所有基因型的8周龄小鼠饲喂对照日粮(CD)或WD,为期20周(图3一).果然,治疗Alb-Cre对照组小鼠WD导致(1)肝脏和体重增加(在线补充图1A-C(2)进行性脂肪变性伴肝内甘油三酯积累(图3 b, (在线补充图1D(3)炎症反应增加,cd45阳性免疫细胞增加,胶原蛋白沉积明显(图3 b - h).然而,重要的是,观察到炎症、cd45阳性细胞募集和胶原沉积的相应增加(图3 b-f)在肝细胞特异性激活核NFATc1的转基因小鼠中(NFATc1一个),且这种损伤在高脂WD饲粮中再次增加。重要的是,与我们在NASH患者中的观察结果一致,我们还测量了带有NFATc1的小鼠血清ALT水平升高一个.驱动的NAFLD进展(在线补充图1E).相反,在肝细胞特异性NFATc1缺乏的小鼠中,疾病进展大大减慢(NFATc1Δ/Δ),因此,NFATc1的消耗减少了肝纤维化和炎症的程度,并几乎完全消除了cd45阳性细胞的招募(图3 b-f).值得注意的是,我们无法观察到NFATc1在肝脂肪变性发展过程中的依赖性,因此转录因子的异位表达和遗传缺失对WD后肝脂肪积累的程度都没有显著影响(图3 g, H,在线补充图1D,F).总的来说,这些发现证实了NFATc1在NAFLD进展中的因果作用,并显示了肝细胞中转录因子的激活,例如,作为高脂肪饮食的结果,会导致明显的炎症和纤维化。

肝细胞中NFATc1的激活驱动肝脏炎症和纤维化。(A)具有肝细胞特异性NFATc1表达的转基因小鼠示意图(Alb-Cre;NFATc1一个.(NFATc1一个.)或删除Alb-Cre;NFATc1Δ/Δ(NFATc1Δ/Δ)和喂养计划,分别为4、12和20周。(B) CD(左)和WD(右)治疗小鼠肝脏切片的H&E分析(n=5)。比例尺= 100µm。cd处理和wd处理20周后肝脏(C-D) CD45、(E-F)苦糖红染色和(G-H)油红-o染色免疫组化分析和定量的代表性图像NFATc1Δ/Δ,Alb-Cre而且NFATc1一个.老鼠(n = 5)。采用双向方差分析进行统计分析,数据以平均值±标准差表示,其中p值为*p<0.05, **p<0.005, ***p<0.0005。
脂毒性脂肪酸诱导肝细胞NFATc1信号激活
游离脂肪酸(FFAs)是肝内甘油三酯池的主要贡献者,最近的研究表明,脂毒性的FFAs在NAFLD的发展和进展中发挥着特别重要的作用。这尤其适用于饱和的FFA棕榈酸盐,它对肝细胞有明显的细胞损伤作用,在NASH患者的血液中浓度特别高。34为了直接测试暴露于棕榈酸盐是否会诱导肝细胞中NFATc1的表达,我们用高浓度的高脂毒性脂肪酸(在线补充图2A而且图4 g).事实上,棕榈酸盐处理导致NFATc1在两种细胞模型中的表达以及mRNA和蛋白质水平显著的剂量依赖性增加(在线补充图2A而且图4模拟).相比之下,与油酸盐孵育,一种非脂毒性的FFA即使在高剂量下也不能诱导原代肝细胞中NFATc1的表达,尽管肝细胞脂肪变性显著积累,如Oil-Red-O染色(在线补充图2A,B).棕榈酸酯和油酸酯是最丰富的游离脂肪酸,占血浆游离脂肪酸总量的一半以上,有研究表明,棕榈酸酯的细胞毒性作用可以通过与油酸酯的共暴露而抵消。与此一致,非脂毒性油酸酯在肝细胞中共同给药抑制了棕榈酸酯对NFATc1的诱导,支持了钙的诱导2 +-调控的转录因子在NAFLD进展中主要是ffa诱导的脂毒性的结果,而不是脂肪堆积本身。脂毒诱导的细胞应激与钙损伤密切相关2 +信号内稳态已经在肝细胞中得到证实。在这里,我们扩展了这些发现,并表明暴露于棕榈酸盐启动了钙的剂量依赖性转移2 +从ER储存到脂肪肝细胞的细胞质(在线补充图2C-E).胞质钙的增加2 +引起各种细胞类型依赖的应激反应通路的激活,35最显著的是NFATc1信号和转录因子。事实上,棕榈酸盐诱导胞质钙的积累2 +与肝细胞中强有力的NFATc1激活相平行,这可以通过反映转录因子低磷酸化活性状态的下带的特别强烈的增加来证明(图4 c, D,在线补充图2A).因此,报告基因分析和免疫荧光染色证实了NFATc1在两种细胞系中对棕榈酸盐处理的核积累和转录活性的增加(图4比;在线补充图2F).

脂毒性脂肪酸导致NFATc1激活。(A - B)用100 μ M(+)和200 μ M(++)棕榈酸盐(pal.)处理12小时后(A)小鼠原代肝细胞和(B) AML12细胞中NFATc1 mRNA的表达。通过qRT-PCR分析NFATc1基因表达,并显示与未处理对照组相比的“相对mRNA水平”。(C - D)用100 μ M(+)和200 μ M(++)棕榈酸酯(pal.)处理12小时诱导(C)小鼠原代肝细胞和(D) AML12细胞中NFATc1蛋白表达。下面的波段代表NFATc1的活跃状态。(E)在肝细胞中进行双荧光素酶报告基因测定Alb-cre验证棕榈酸盐诱导的NFATc1的转录激活。将NFAT响应启动子荧光素酶报告子与空载体或NFATc1野生型(NFATc1wt)表达载体,随后用200 μ M棕榈酸酯(pal.)处理24小时。(F) AML12细胞中的NFATc1免疫荧光显示,在200 μ M棕榈酸盐处理12小时后,转录因子发生核易位。比例尺= 100µm。(G) AML12细胞中棕榈酸诱导的NFATc1核定位的定量分析。采用单因素方差分析(A,B)、双向方差分析(E)和无配对t检验(G)进行统计分析。数据用平均值±SD表示,*p≤0.05,**p≤0.005,****p≤0.0001。
NFATc1促进肝脏内质网应激诱导的UPR和免疫信号转导
为了探索NFATc1调节NAFLD进展的潜在机制,我们接下来研究了NFATc1依赖的基因表达,并在AML12细胞中进行了转录组分析。为此,我们从(ha标记的)c.n.NFATc1或对照质粒转染的AML12细胞中提取mRNA,并进行RNA-Seq分析。对于转染控制,我们进行了western blot和qRT-PCR分析(图5一个).PCA显示了nfatc1诱导的和对照的不同聚类(在线补充图3A).RNA-Seq分析显示有636个差异表达基因,其中471个基因在NFATc1核激活后上调(图5 b).此外,反应组通路分析进一步表明,最显著富集的通路参与干扰素和细胞因子/趋化因子信号、细胞死亡调节或细胞应激反应(图5 c).具体来说,NFATc1的激活导致了高度显著的和可复制的促炎细胞因子的诱导(如,Ccrl2和Ccl5)和趋化因子(例如,Cxcl2, Cxcl9 Cxcl10, Cxcl11),炎症转录因子(如,Stat1和Stat2)和细胞死亡标记基因(如Bak1,Casp7和Tnfsf10)(图5 d).值得注意的是,我们确认了nfatc1依赖的炎症细胞因子和趋化因子调控,例如,IL-1β, Ccl5, Cxcl9, Cxcl10而且Cxcl11在小鼠NAFLD进展模型中(在线补充图3B,C).

NFATc1调节基因特征和信号机制。(A)代表性的western blot和qRT-PCR显示NFATc1成功转染并在AML12细胞中表达。(B)在AML12细胞中NFATc1过表达的RNA-Seq分析中,描述显著差异表达基因z分数的热图。(C)反应组路径分类分析,显示RNA-Seq识别的AML12细胞中最显著调控的NFATc1基因签名(log2倍值≥0.5/≤−0.5;p≤0.05;基本意思> 10)。(D) AML12细胞中NFATc1激活差异调控候选基因的qRT-PCR验证。数据以均值±SD表示,p值为*p<0.05, **p<0.005。统计分析采用非配对t检验。
最重要的是,我们的转录组分析还发现了NFATc1激活和内质网应激通路诱导,特别是终端PERK(蛋白激酶rna样内质网激酶)UPR之间强大且高度可复制的联系。事实上,我们发现了多种信号成分的显著(20倍)诱导,例如,Eif2ak2,Atf3和dit3(剁)对NFATc1活化(图5 c, D).内质网腔内蛋白质稳态紊乱后,激活三种典型的ER应力传感器蛋白ATF6、IRE1和PERK,可诱导UPR。在激活时,UPR信号控制多种细胞机制,以减少蛋白质合成和增加蛋白质折叠能力。然而,虽然生理UPR信号可以使细胞维持细胞内稳态,但过度的UPR激活会导致病理变化,如细胞损伤和死亡。例如,研究表明,脂毒性细胞损伤(如棕榈酸盐)可导致慢性内质网应激感应和UPR信号,并在NAFLD进展中引发细胞死亡和炎症。36-38这对于通过Eif2α-ATF4-CHOP途径的PERK激酶驱动的信号传递尤其如此。36 38 39例如,高水平的PERK和CHOP在NASH患者中被发现,特别是在那些有不良病程的患者中。40在这里,我们发现NFATc1的激活确实可以诱导PERK激酶信号,但也可以诱导PKR的磷酸化,PKR是eif2α激酶家族的另一个成员,参与细胞应激反应和UPR信号(图6).具体来说,NFATc1的激活——无论是基因上的还是棕榈酸盐刺激后引起的PERK (pPERK)、PKR (pPKR)和Eif2α (pEif2α)磷酸化的增加,以及随后核心下游信号元件(例如CHOP)的诱导,均在培养的AML12细胞和原代肝细胞中均存在(在线补充图4A,图6 a e).在服用了wd的小鼠中也发现了类似的结果(图6 f).

核NFATc1促进末端未折叠蛋白反应(UPR)信号通路。(A)免疫印迹检测显示,单独暴露于棕榈酸盐(+=200 μ M) 12小时或结合敲除NFATc1 (scinfatc1) 12小时后,AML12细胞中NFATc1、pPERK、PERK、pPKR、PKR、ATF4、TRB3、p-Eif2α、Eif2α和CHOP蛋白水平。(B)单独用棕榈酸酯(200 μ M)处理或与scaminc1联合处理AML12细胞中pPERK/PERK和pPKR/PKR的密度图。(C)从NFATc1表达差异的转基因小鼠中分离原代肝细胞和随后的棕榈酸盐处理示意图。(D)代表性的western blot显示原代肝细胞中nfatc1依赖性的pPERK、PERK、pPKR、PKR、ATF4、TRB3、p-Eif2α、Eif2α和CHOP蛋白水平。(E)将原代小鼠肝细胞暴露于棕榈酸盐(+=100 μ M, ++=200 μ M)中12小时,用免疫印迹法分析pPERK、PERK、pPKR、PKR、ATF4、TRB3、p-Eif2α、Eif2α和CHOP水平的变化。(F)利用cd处理和dd处理的转基因小鼠(GEM)模型的肝组织裂解液进行Western blot分析,以确定脂肪诱导和nfatc1依赖性的pPERK、PERK、pPKR、PKR、ATF4、TRB3、p-Eif2α、Eif2α和CHOP的表达。
此外,NFATc1沉默后,末端UPR反应严重受损(图6 a e, (在线补充图4B-D)在体外和小鼠NAFLD进展模型中,甚至在长时间暴露于WD (图6 f).值得注意的是,NFATc1的激活似乎与肝细胞中其他两个内质网应激传感器(IRE1/XBP1和ATF6)的激活无关,因此我们无法观察到NFATc1依赖的表达差异(在线补充图4E,F)关于棕榈酸盐治疗。这一观察结果也与之前的报道一致,尽管IRE1/XBP1和ATF6通路也可以上调CHOP,但PERK通路通过选择性上调ATF4转译在NAFLD进展中占主导地位,随后诱导CHOP转录促进细胞死亡。3 41
总之,这些研究为在进行性NAFLD中脂毒脂肪酸诱导的肝细胞NFATc1激活和诱导有害的PERK/PKR-CHOP UPR通路之间的机制联系提供了令人信服的证据。
NFATc1缺失保护肝细胞免受er应激诱导的炎症小体激活和凋亡
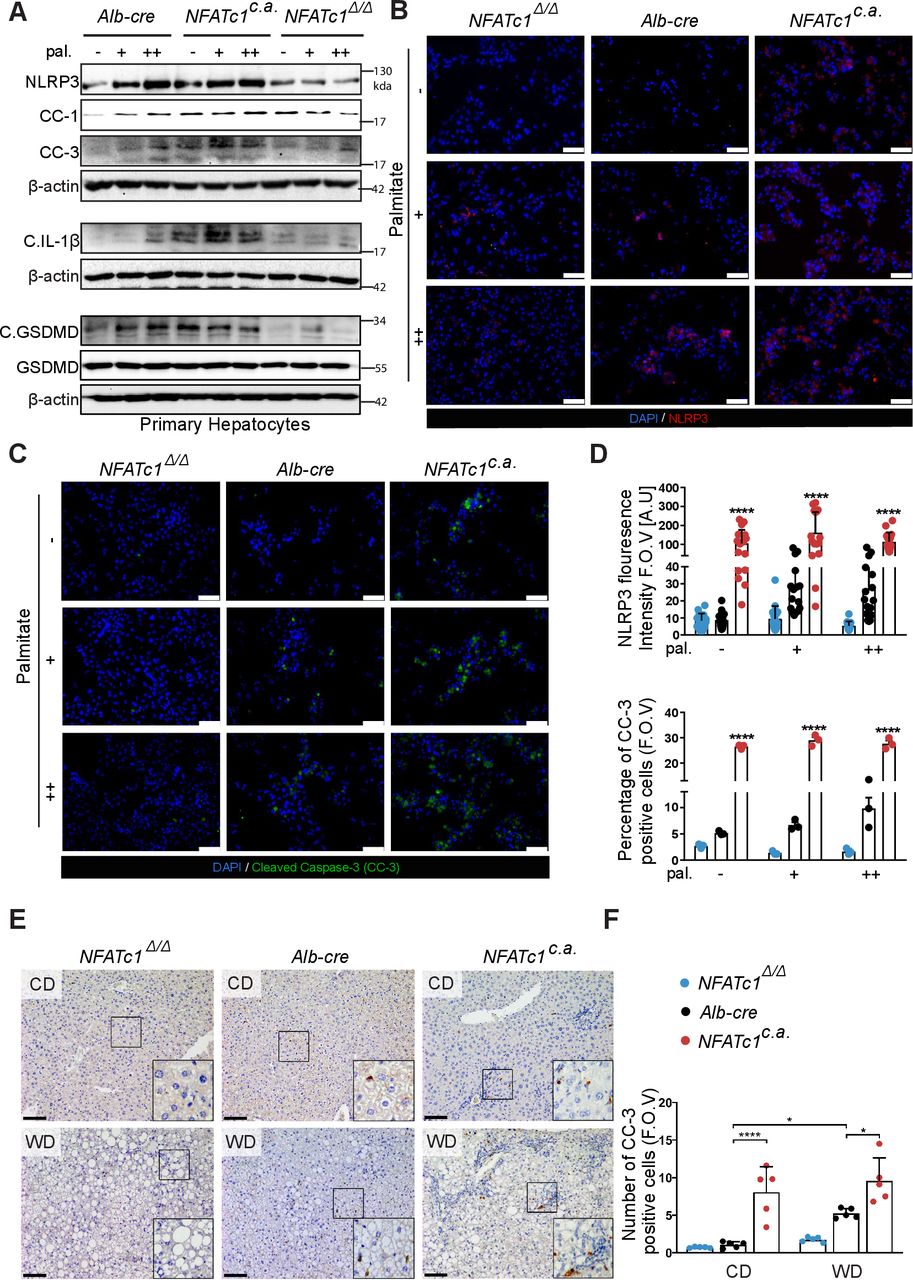
最近的研究明确表明,慢性内质网应激通过PERK-CHOP信号通路促进细胞凋亡,并激活含有3 (NLRP3)炎症小体的nod样受体家族pyrin结构域。37NLRP3是一种多聚蛋白复合物,刺激caspase-1依赖的白细胞介素-1β (il -1β)和gasdermin-D的裂解,以诱导炎症和细胞死亡的促炎症形式,称为焦亡。在这里,我们研究了在进展性NAFLD中,终端PERK-CHOP信号激活是否促进肝细胞死亡和NLRP3驱动的焦亡激活,如果是,这是否依赖于NFATc1。为此,我们分析了内质网应激诱导的NLRP3炎症小体激活以及caspase-1介导的IL-1β和gasdermin在原代肝细胞、AML-12细胞和NFATc1表达差异的转基因小鼠肝脏中的裂解。western blot、免疫荧光和免疫组化分析证实,脂毒诱导的UPR信号通路促进细胞凋亡(由切割的caspase-3指示)、NLRP3炎症小体诱导的细胞因子释放(由切割的IL-1β指示)和细胞焦亡(由切割的caspase-1指示和增加的gasdermin-D激活(C.GSDMD)) (图7 f,在线补充图4G,H).重要的是,rpu驱动的炎症小体激活和细胞死亡启动需要NFATc1的存在,因此,转录因子的遗传缺失阻止了棕榈酸诱导的caspase-3激活,并阻止了nlrp3驱动的焦亡和IL-1β裂解,在体外和NAFLD进展模型中(图7 f,在线补充图4G,H).相比之下,与我们的研究结果相一致在线补充图2在原代肝细胞中,非脂毒性油酸盐治疗既没有诱导NFATc1激活,也没有诱导末端UPR信号,也没有诱导随后的nrlp3 -炎症体诱导的细胞死亡(在线补充图5).

nfatc1依赖的细胞死亡和体内外炎症小体激活。(A)代表性免疫印迹显示,在棕榈酸盐处理12小时(+=100 μ M和++=200 μ M)后,原代肝细胞NLRP3、裂解caspase-1 (CC-1)、裂解caspase- 3 (CC-3)、裂解白介素(IL)-1β、裂解GSDMD和GSDMD (Gasdermin D)的nfatc1依赖性变化。(B)棕榈酸盐处理后小鼠原代肝细胞NLRP3和(C) CC-3的免疫荧光分析。比例尺= 100µm。(D)图表示NLRP3的荧光强度和CC-3阳性肝细胞(F.O.V)的百分比。(E)治疗20周小鼠肝脏切片CC-3染色免疫组化分析。比例尺= 100µm。(F) CC-3阳性肝细胞定量分析(F.O.V)。数据以平均值±标准差表示;p值为*p<0.05, ****p<0.0001。 Statistical analysis was performed using one-way analysis of variance (ANOVA) (D) and two-way ANOVA (F). CD, control diet; WD, western diet.
总的来说,这些在原代肝细胞和转基因小鼠中进行的实验强调了核NFATc1在驱动内质网末端应激反应促进NAFLD进展中的关键作用。
抑制慢性内质网应激反应可减弱nfatc1诱导的NAFLD进展
基于这些结果,我们测试了内质网应激的药物抑制是否可以阻止nfatc1引发的NAFLD疾病进展。为此,我们分析了牛磺酸去氧胆酸(TUDCA)对原代肝细胞和AML12细胞以及WD喂养的转基因小鼠中nfatc1依赖机制和细胞功能的影响,TUDCA是内质网应激反应的成熟抑制剂。图8,在线补充图6A-C).
的结果一致图6和7在体外和体内,通过棕榈酸盐治疗或c.n.NFATc1转染诱导内质网应激可导致末端UPR信号(如CHOP)激活,并随后诱导NRLP3炎症小体效应通路。然而,有趣的是,TUDCA的应用充分阻断了内质网应激诱导的末端UPR信号应答和炎症小体激活。具体而言,我们观察到终端UPR的显著封锁(表明CHOP表达的丢失),细胞死亡诱导的抑制(通过切割caspase-3反映)和nrlp3 -炎症小体介导的细胞因子释放(如IL-1β)的失活,即使在活性NFATc1 (图8 a, B,在线补充图6A).在NAFLD进展模型中也证实了类似的效果,在该模型中,tudca介导的对末端UPR信号的抑制和随后的NLRP3激活与炎症的大幅减少、cd45阳性免疫细胞的招募和肝纤维化相关,尽管表达了本构活性NFATc1 (图8 d - i,在线补充图6D-E).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
牛磺酸去氧胆酸(TUDCA)可减弱进展性非酒精性肝病(NAFLD)中nfatc1依赖的未折叠蛋白反应(UPR)信号通路诱导的炎症和纤维化。(A)免疫印迹显示CHOP、NLRP3和CC-3蛋白表达Alb-cre与对照组处理的细胞相比,单独用棕榈酸盐(++=200 μ M)处理或与TUDCA浓度增加(100-500 μ M)处理的原代肝细胞。(B)在NFATc1构成激活的AML12细胞中,在500 μ M TUDCA存在或不存在的情况下,评估CHOP、NLRP3和CC-3的蛋白水平。转染scinfatc1的细胞作为对照。(C)预防治疗方案示意图。(D)免疫印迹显示治疗20周后肝组织裂解液中CHOP和NLRP3蛋白水平Alb-cre老鼠和NFATc1一个.老鼠。(E) H&E染色及免疫组化分析(F) CD45和(G)苦糖红染色。比例尺= 100µm。(H)图表示CD45阳性细胞的百分比和(I)苦甘草红染色面积的百分比(F.O.V)。数据以均值±SD表示,p值为*p<0.05, **p<0.005, ***p<0.0005, ****p<0.0001。统计分析采用双向方差分析。
总之,这项研究有力地支持NFATc1在NAFLD进展中的重要作用,并证明该功能是基于调节慢性内质网应激反应和随后NLRP3炎症小体激活。此外,我们提供的证据表明,内质网应激反应的药理学抑制(如通过TUDCA)可以克服nfatc1驱动的疾病进展。
讨论
本研究旨在破译NAFLD进展的关键分子机制,从而为开发新的治疗策略提供合理的基础。因此,我们将重点放在钙响应型NFAT转录因子家族上,它控制着炎症相关和代谢疾病的大量细胞过程,例如胰岛素抵抗和糖尿病、肥胖和癌症。12 14 17 18 20在这里,我们发现了NFATc1在NAFLD向NASH发展过程中的重要作用。因此,NFATc1表达和核定位在健康肝脏中较弱甚至缺失,但在进展性NASH中强烈诱导。此外,用脂肪酸(例如棕榈酸或WD)处理可以诱导NFATc1在原代和已建立的肝细胞以及小鼠肝组织中的表达、核易位和转录活性。此外,小鼠肝脏中NFATc1核的肝细胞特异性激活——要么在WD喂养后,要么通过转录因子(NFATc1)的遗传诱导一个) -可加速肝损伤,表现为组织炎症增加、cd45阳性细胞聚集和肝纤维化的进行性形成。这些结果,以及观察到NFATc1的肝细胞特异性遗传缺失可以防止脂肪酸诱导的非酒精性脂肪肝的进展超过肝脏脂肪变性阶段,为Ca的关键作用提供了强有力的实验证据2 +反应性转录因子在进展性NAFLD中的作用
机制上,NFATc1通过肝细胞内不受限制的内质网应激信号应答促进NAFLD加速。ER负责正确的蛋白质折叠,3 42研究表明,钙稳态或内质网蛋白质折叠机制的缺陷会导致内质网应激和随后UPR通路的激活。UPR通路协调了许多修复内质网应激的关键调节机制,包括抑制蛋白质合成和加速内质网蛋白质降解。一般来说,UPR的激活足以修复瞬态和轻度的内质网应激。3 37 43-46然而,如果内质网应激是严重的和未解决的,它可以导致持续激活的UPR信号通路,最终导致细胞死亡,炎症小体激活和加速器官损伤。最近的研究表明,脂肪积累可触发慢性内质网应激,并随后激活肝细胞末端UPR信号级联。37 11 37 47 48在这里,我们第一次展示了Ca的基本作用2 +内质网应激诱导的NAFLD进展中的响应性转录因子NFATc1。具体来说,长时间接触脂肪酸可诱导体外和进行性NAFLD小鼠肝细胞中NFATc1的表达、核定位和活性。机制上,NFATc1通过末端的PERK-CHOP信号促进内质网应激感知,随后诱导NLRP3激活,49-51参与细胞死亡起始和炎症的大分子炎症小体复合体。7 36 38 48末端PERK-CHOP通路在慢性内质网应激驱动的NLRP3炎性小体激活和凋亡中的显著作用已在各种疾病中得到证实,特别是在与代谢紊乱相关的疾病中。47 48 52 53例如,在NAFLD中,CHOP激活诱导NRLP3炎症小体复合体,以促进肝细胞死亡和炎症反应,以应对未解决的内质网应激。3 7 36我们的研究结果不仅证实了这些先前的观察结果,而且还证明了NFATc1的激活对于通过PERK-CHOP分支的脂肪诱导内质网应激信号以及随后NRLP3炎症小体通路的激活是必需的。因此,即使在高脂肪饮食的长期刺激下,NFATc1的遗传缺失也会阻止肝细胞在原代肝细胞和肝组织中的末端UPR信号、NRLP3激活和细胞死亡启动。
最后,我们评估了抑制慢性内质网应激信号是否可以保护脂肪肝免受nfatc1诱导的疾病进展。为此,我们将长时间的高脂肪刺激与TUDCA的应用相结合,TUDCA是一种天然亲水性胆汁酸和牛磺酸熊去氧胆酸(UDCA)的结合物,它被食品和药物管理局批准用于治疗原发性胆道胆管炎(PBC)。最近的多中心随机临床试验表明,TUDCA治疗PBC的安全性和耐受性与UDCA相同,甚至可能更好地缓解疾病症状,提示牛磺酸结合物治疗PBC的有效性更高。54TUDCA对胆汁淤积性疾病的疗效主要归因于其通过增加胆汁流量和胆汁酸分泌对肝细胞的胆汁抑制和细胞保护作用。55重要的是,许多临床前研究也证明了TUDCA在非胆汁淤滞性肝病,特别是NAFLD中具有显著的治疗潜力,它通过减轻内质网应激和阻断末端PERK-CHOP信号传导的能力,发挥了强大的细胞保护作用。7 36 56我们的研究结果有力地支持了这些努力,并为TUDCA在预防NAFLD进展方面的有效性提供了机制基础。我们发现,TUDCA的应用通过抑制nfatc1介导的内质网应激感应和末端UPR信号激活,有效地干预了疾病的加速。TUDCA治疗可以阻断nfatc1诱导的CHOP-NRLP3炎症小体的激活,从而降低肝脏的炎症、凋亡和纤维化程度,即使是在长期高脂肪饮食中也是如此。
综上所述,本研究确定钙信号响应转录因子NFATc1在NAFLD进展中起关键作用。我们发现NFATc1通过促进慢性内质网应激反应和激活NRLP3炎症小体来驱动脂肪诱导的NASH。这项研究不仅有助于更好地理解NFATc1在NAFLD中的信号,而且为近期旨在通过药物干预慢性内质网应激反应来预防疾病进展的临床试验提供了机制基础。
数据可用性声明
资料应合理要求提供。所有与研究相关的数据都包含在文章中或作为补充信息上传。
伦理语句
病人同意发表
伦理批准
所有动物实验均经当地动物护理和使用委员会(LAVES)批准,并按照欧洲实验动物科学协会联合会(FELASA)的规定进行。
致谢
我们非常感谢巴基斯坦高等教育委员会为Muhammad Umair Latif提供博士奖学金。我们感谢陈乃明博士对项目发展的投入,感谢Sarah L. Hanheide对小鼠育种的贡献。我们感谢来自Goettingen NGS综合基因组学核心单元的Gabriela Salinas对样本的测序。所有的示意图都是通过BioRender.com制作的。最后,我们感谢所有实验室成员的深刻讨论。
参考文献
脚注
推特@ShivKSingh6
贡献者MUL完成了大部分的实验,分析和解释了结果。GES进行了文库准备、rna序列分析和图形设计。RR有助于RNA-seq数据的生物信息学分析。SM负责小鼠的繁殖和处理。CSG和IS-T在AML12细胞中进行钙含量测定实验,AR在原代肝细胞中进行。KR协助制定实验计划并进行实验。所有活体实验的批文都是EH写的。SKS提供了智力投入,并参与了论文的图形设计。AM有助于优化原代肝细胞分离程序。UJB为IHC分析和稿件校对提供支持。 PS provided human tissues as well as helped in analysing the IHC staining. SCB and HB provided human NASH patient samples. AN provided scientific input in planning experiments and data interpretation. IB provided scientific inputs for this study and helped in planning experimental models. VE was the principal investigator of the study and was responsible for study concept and design. Together with MUL, he was also responsible for manuscript writing. VE is the guarantor and responsible for the overall content of the study.
资金该项目主要由DFG (KFO-5002)资助。此外,它还得到了德国癌症援助Shiv K. Singh (70112999;Max-Eder组),到Albrecht Neesse (70113213;Max-Eder集团)和Eisabeth Hessmann (70112108), DFG赠款给Shiv K. Singh和Elisabeth Hessmann (KFO-5002), DFG赠款给Ivan Bogeski (SFB1190 P17)和大众基金会/下萨克森州文化和科学部(MWK)给Volker Ellenrieder(11-25 76251-12-3/16)。
相互竞争的利益没有宣布。
来源和同行评审不是委托;外部同行评议。
补充材料本内容由作者提供。它没有经过BMJ出版集团有限公司(BMJ)的审查,也可能没有经过同行评审。讨论的任何意见或建议仅仅是那些作者(s)和不被BMJ认可。BMJ放弃从放在内容上的任何依赖产生的所有责任和责任。如果内容包含任何翻译材料,BMJ不保证翻译的准确性和可靠性(包括但不限于当地法规、临床指南、术语、药品名称和药物剂量),并且不对翻译和改编或其他原因引起的任何错误和/或遗漏负责。
请求的权限
如果您希望重用这篇文章的任何部分或全部,请使用下面的链接,它将带您访问版权清除中心的RightsLink服务。您将能够快速获得价格和以多种不同方式重用内容的即时许可。