条文本

摘要
客观的赖氨酸氧化酶样蛋白2 (LOXL2)有助于不同肿瘤实体的肿瘤进展和转移,但其在胰腺导管腺癌(PDAC)中的作用尚未在具有免疫能力的体内PDAC模型中得到评价。
设计为此,我们使用PDAC患者数据集、患者衍生的体内和体外异种移植模型和四种条件基因工程小鼠模型(GEMMS)来分析LOXL2在PDAC中的作用。GEMM-based研究k -+ / LSL-G12D;Trp53LSL-R172H;Pdx1-Cre和k -+ / LSL-G12D;Pdx1-Cre小鼠(KC)进行杂交Loxl2等位基因floxed小鼠(Loxl2Exon2fl / fl)或条件Loxl2overexpressing老鼠(R26Loxl2KI /吻)以生成KPCL2KO或KCL2KO和KPCL2KI或KCL2KI用于研究总体生存率的小鼠;肿瘤发病率、负担与分化;转移;上皮向间质转化(EMT);茎干和细胞外胶原基质(ECM)组织。
结果使用这些PDAC小鼠模型,我们显示whileLoxl2消融对原发肿瘤的发育和生长几乎没有影响,它的消失显著减少了转移和增加了总生存率。我们将这种效应归因于非细胞自主因素,主要是ECM重塑。Loxl2另一方面,过表达促进了原发性和转移性肿瘤的生长,降低了总生存率,这可能与EMT和stemness的增加有关。我们还确定了肿瘤相关的巨噬细胞分泌的抑癌素M (OSM)作为LOXL2表达的诱导剂,并显示了在体内靶向巨噬细胞的影响Osm而且Loxl2表达和胶原纤维排列。
结论综上所述,我们的研究结果建立了LOXL2在PDAC中的新的病理生理作用和功能,可能被用于治疗转移性疾病。
- 胰腺癌
- 细胞基质的相互作用
- 巨噬细胞
- 分子肿瘤学
- 胰腺纤维化
数据可用性声明
本文中使用的数据集的数据可在一个公共的、开放的存储库中获得。其他资料应合理要求提供。https://www.nature.com/articles/ng.3398;https://bmccancer.biomedcentral.com/articles/10.1186/s12885-016-2540-6;//www.marcconsult.com/content/66/9/1665。
这是一篇开放获取的文章,按照创作共用署名非商业性(CC BY-NC 4.0)许可发布,该许可允许其他人以非商业性的方式发布、混编、改编、构建本作品,并以不同的条款授权他们的衍生作品,前提是原创作品被正确引用,给予适当的荣誉,任何更改都被注明,且使用是非商业性的。看到的:http://creativecommons.org/licenses/by-nc/4.0/。
来自Altmetric.com的统计
本研究的意义
关于这个问题,我们已经知道了什么?
胰腺导管腺癌(PDAC)肿瘤微环境的特征之一是广泛的粘连增生反应,包含密集的纤维组织和丰富的富含胶原的细胞外基质(ECM),可以调节肿瘤的形成、进展和化疗耐药。
赖氨酸氧化酶家族的蛋白质,包括赖氨酸氧化酶样蛋白2 (LOXL2),负责ECM胶原蛋白和弹性蛋白交联,导致调节肿瘤进展的生化、物理和机械效应。
LOXL2已被证明可以通过细胞自主机制,如激活上皮向间质转化(EMT)和非细胞自主机制,如ECM组织,增强肿瘤的进展、侵袭和转移。
本研究的意义
新的发现是什么?
肿瘤相关巨噬细胞(TAM)分泌的oncostatin M (OSM)作为PDAC肿瘤LOXL2表达的诱导物。
的损失Loxl2在喀斯特G12D驱动型PDAC小鼠肿瘤通过非细胞自主因子(主要是ECM胶原重塑)显著减少转移并增加总生存率。
过度的Loxl2在喀斯特G12D驱动型PDAC小鼠肿瘤促进原发性和转移性肿瘤生长,并通过增加EMT和茎干降低总生存率。
体内靶向TAMs减少Osm而且Loxl2表达,导致转移减少。
在可预见的未来,它会对临床实践产生怎样的影响?
LOXL2在PDAC肿瘤发展中的靶向作用方式和时间可能具有不同的意义和生物学效应。
LOXL2抑制剂与TAMs或osm介导的信号传导抑制剂联合使用可能协同降低PDAC肿瘤细胞的转移潜力。
简介
胰导管腺癌(PDAC)是世界上最致命的实体肿瘤之一,也是癌症相关死亡最常见的原因之一。1 2一般来说,长期以来,PDAC患者的治疗都是类似的,没有分层的治疗方案,这很大程度上是因为人们一致认为不存在分类/分子亚型。然而,这一教条在过去10年的大量转录组研究中受到了挑战,尽管不同研究中确定的亚型数量有所不同,但始终存在两种亚型:(1)祖细胞/经典型和(2)准间充质/鳞状细胞/基底型。3.
虽然这些分子图谱还没有影响临床治疗决策,但它们已经加强了肿瘤微环境(TME)在PDAC中的作用。4 - 6PDAC的一个特征是复杂的多细胞TME的发展,具有广泛的粘连增生反应,其特征是致密的纤维组织7可以对PDAC细胞产生显著的生化、物理和机械影响,8以及丰富的细胞外基质(ECM),由胶原蛋白、纤维连接蛋白、透明质酸和腱蛋白C等组成。9ECM主要通过增加胶原沉积、交联和纤维排列来调节肿瘤的硬度和组织。虽然后者与PDAC肿瘤进展、化疗耐药和转移有关,10 - 13PDAC中基质和ECM(即胶原沉积)的促肿瘤作用最近受到了挑战,14日15强调了我们对PDAC基质的不完全了解。
为此,我们开始评估ECM组织在PDAC中的作用。ECM胶原和弹性蛋白的交联主要由赖氨酸氧化酶(LOX)家族蛋白催化,该蛋白家族由5个成员组成(LOX和4个相关酶,赖氨酸氧化酶样蛋白1 (LOXL1)-4)。16 - 19LOX和LOX样蛋白,特别是LOXL2,已经在许多肿瘤实体中进行了研究;然而,目前还没有确定LOX或特定的LOX样蛋白如何促进肿瘤进展、侵袭和转移的通用作用机制,在18到22岁表明肿瘤特异性以及细胞自主和非细胞自主因素可能起作用。
这些挑战阻碍了LOX和LOX样蛋白抑制剂向临床的转化,但同时也强调了仍需要进一步研究这些蛋白在肿瘤特异性环境中的作用。因此,由于PDAC中胶原纤维组织的促肿瘤作用,以及我们之前对LOXL2在其他肿瘤实体中的作用的分析研究,汽车出行我们利用了PDAC患者数据集、人类患者衍生的异种移植模型和我们之前发表的研究成果Loxl2敲除和过表达小鼠模型,解剖LOXL2在PDAC肿瘤起始、进展和转移中的作用。
方法
基因表达数据集,GSEA分析,Kaplan-Meier分析和Pearson相关
本研究中使用的基因表达数据集是公开的。来自Janky的数据集等28从GEO (GSE62165)下载;莫菲特的数据集等4从GEO (GSE71729)下载;元数据集,包含数据集GSE15471, GSE16515, GSE22780和GSE32688,如ref中所述生成29。
PDAC patient-derived异种移植
根据与国家调查中心Oncológicas (CNIO)的材料转移协议,Manuel Hidalgo博士获得了先前建立的PDAC患者源性异种移植组织(PDAC PDX)(参考编号:CNIO)。I409181220BSMH)。如前所述,在6-8周龄NU-Foxn1nu雌性裸鼠(Envigo, Spain)中扩展指示的PDXs。30.PDXs 110314、140114和010414来自慕尼黑工业大学伊萨尔医院切除的原发性患者PDAC肿瘤,随后在慕尼黑工业大学建立。
细胞培养
如前所述,建立人Panc185、Panc215、Panc253、Panc010414和Panc354 PDAC pdx衍生培养。30.建立KPC文化已经在前面描述过了31细胞系使用唯一标识符编号(如ID6, ID11, ID85, ID95)。KPCL2KO和KPCL2KI细胞系是从外植的肿瘤中建立的。简单地说,肿瘤被切碎,用胶原酶(胶原酶型P, Cat no。J62406.03, Alfa Aesar),随后在RPMI 1640培养基(Invitrogen, Cat no。61870044)含有10%胎牛血清(Invitrogen)和50单位/mL青霉素/链霉素(Invitrogen,猫号;11548876)。上皮克隆被挑选,聚集,进一步扩大到一个异质癌细胞系,重新基因分型如所述26并使用唯一标识符编号(如ID11, ID32, ID86)。
基因改造小鼠模型
PDAC小鼠模型:k -+ / LSL-G12D;Trp53LSL-R172H;Pdx1-Cre小鼠模型(KPC)和k -+ / LSL-G12D;Pdx1-Cre小鼠模型(KC)之前已经描述过,32并保存在C57BL/6的背景中。的液氧Loxl2等位基因(外显子2)小鼠Loxl2fl / fl和条件Loxl2-EGFP过表达小鼠模型R26Loxl2-EGFPKI /吻已经被描述过了,26他们有不同的背景。在进一步的PDAC研究中,肿瘤小鼠由雄性KPC小鼠与雌性KPC小鼠杂交产生Loxl2Exon2fl / fl产生杂合子(Het) KPCL2KO老鼠(KPCL2+ /−)或与雌性R26Loxl2KI /吻小鼠产生Het KPCL2KI老鼠(KPCL2+ /吻).后代的出生符合预期比率和KPCL2KO小鼠未见视觉功能缺陷或胰腺组织学异常(数据未显示)。将不同品种的Het小鼠进一步杂交得到KPCL2+/+, KPCL2+ /−和KPCL2−−/老鼠的Loxl2研究或获得KPCL2+/+, KPCL2+ /吻, KPCL2KI /吻老鼠的Loxl2超表达研究。使用的基因分型引物列于在线补充表S2。两个KPCL2的野生型(+/+)窝伴侣KO和KPCL2KI在所有实验中,系均作为对照。为了进行肿瘤负荷和转移分析,小鼠在出生后17-18周被处死(队列2)。对器官、肿瘤和宏观转移进行分离/测定、称重和数字记录。将组织固定在4%多聚甲醛中,快速冷冻或如上所述消化,以建立原代细胞系。在生存曲线分析中,只有在人道终点牺牲的小鼠和发生胰腺肿瘤的小鼠被纳入(队列1),其他所有小鼠都被排除在分析之外。
同时还采用了一种较为温和的PDAC模型。在这些研究中,雄性KC小鼠与雌性KC小鼠交配Loxl2Exon2fl / fl生成Het KCL2KO老鼠(KCL2+ / -)或与雌性R26Loxl2KI /吻小鼠产生Het KCL2KI老鼠(KCL2+ /吻).将不同品种的Het小鼠进一步杂交得到KCL2+/+, KCL2+ /−和KCL2−−/老鼠的Loxl2研究或获得KCL2+/+, KCL2+ /吻和KCL2KI /吻老鼠的Loxl2超表达研究。两个KCL2的野生型(+/+)凋落物KO和KCL2KI在所有实验中,系均作为对照。为了进行肿瘤负担和转移分析,队列2小鼠在出生后38-40周被处死,动物处理如上所述。对于生存曲线分析,只包括在人道终点牺牲的小鼠(队列1)。
肿瘤形成动力学的差异以及野生型KPCL2表现出转移的小鼠百分比的差异KO和KPCL2KI和KCL2KO和KCL2KI观察到的线条,并确定是由于背景差异。
患者和公众参与研究
患者和公众都没有直接参与这项研究。其中包括通过生物银行医院Ramón y Cajal-IRYCIS (PT13/0010/0002)在知情同意的情况下提供的患者样本,该样本已纳入西班牙国家生物银行网络(ISCIII生物银行注册编号为ISCIII)。B.0000678)(详见补充方法)。
统计分析
除非另有说明,结果以均数±SEM表示。采用单因素方差分析与Tukey或Dunnett后验进行两两多重比较。除非另有说明,未配对双侧(CI为95%)使用学生t检验来确定两组均值之间的差异。P值<0.05认为有统计学意义。所有分析均使用GraphPad Prism V.6.0进行。
其他方法可以在在线补充材料。
结果
LOXL2信使RNA表达与PDAC患者的总生存率差和上皮向间质转化相关
的转录水平LOXL2首先在三个公开的转录组数据集GSE62165中评估表达,28元数据集29GSE71729,4和之前的出版物一样,到三十五LOXL2与邻近正常组织相比,整个胰腺肿瘤样本中的表达显著升高(图1一个).在之前发表的一系列文章中,这种增加在信使RNA (mRNA)和蛋白质水平上得到了独立验证31新切除的PDAC肿瘤(图1 b而且在线补充图S1A,B).关于生存,观察到中位总生存(OS)有明显的偏差和下降LOXL2-高表达患者(50%)与LOXL2 -贝利的低表达患者(50%)等系列36(图1 c).此外,LOXL2与其他三种亚型相比,更具侵袭性的鳞状细胞亚型表达更高(图1 d),由贝利分类等。36基因集富集分析(GSEA)比较了基因组中上、下四分位的样本LOXL2对上述三个数据集进行了表达,我们观察到在所有系列中显著且普遍富集的通路(在线补充图S2A),上皮向间充质转化(EMT)和转化生长因子β (TGFß)信号一致且显著增强LOXL2高表达患者,独立于所分析的数据集(图1 e而且在线补充图S2A,B).EMT中,对患者上皮和间充质基因的Spearman秩序相关或Pearson相关矩阵进行排序LOXL2mRNA水平与受监督的层次聚类(欧氏距离测量,平均连锁聚类)证实呈正相关LOXL2EMT和间充质基因的mRNA表达量(在线补充图S3A而且图1 f).同样,高水平的LOXL2生存期和间隔参数明显较差(在线补充图S3B).

LOXL2mRNA表达与PDAC患者的总生存率和EMT较差相关。(A)差异表达LOXL2在GSE62165、META数据集和GSE71729中,邻近(形容词)正常组织与PDAC肿瘤和转移(met)的比较。学生t检验。(B)LOXL2在手术切除的人类PDAC肿瘤(n=25)和4个正常胰腺(nPanc)对照组(左)中,相对mRNA水平±SD (n=2个技术重复)。合并平均±扫描电镜分析包括4个原发性KPC肿瘤(右)。(ns,不重要;* * * * p < 0.0001;双面t检验+ Mann-Whitney U检验)。(C)贝利(n=96)数据集PDAC患者的总生存期,根据的中位数分层LOXL2表达式。HR=风险比,Cox比例风险回归模型。生存分析采用Log-rank检验。(D)差异表达LOXL2在PDAC肿瘤中,亚型分为祖细胞、鳞状细胞、免疫原性或来自贝利的ADEX等数据集。(**P<0.01, **** P< 0.0001, Dunnett后验单因素方差分析)。(E)来自转录组学分析(GSE62165, META和GSE1729数据集)的EMT途径富集图LOXL2高和LOXL2低的病人。罗斯福< 0.25。(F)筛选出的179例人PDAC (TCGA)患者间充质相关和上皮相关基因的Pearson相关矩阵LOXL2mRNA水平和最近的邻居。对矩阵进行监督层次聚类(欧氏距离测量、平均连锁聚类)。EMT,上皮向间质转化;FDR,错误发现率;LOXL2,赖氨酸氧化酶样蛋白2;信使RNA信使RNA;PDAC,胰导管腺癌;TCGA,癌症基因组图谱。
巨噬细胞通过OSM在pdx来源的PDAC细胞中诱导LOXL2和EMT
接下来,我们分析了一组PDAC PDX肿瘤中的LOXL2蛋白水平作为模型系统。在分析的11个PDX肿瘤中,只有高度转移的265个PDX模型37表示LOXL2 (图2一个),这表明异种移植组织要么在体内传代过程中丢失LOXL2,要么在小鼠中缺乏介导LOXL2表达的人类特异性TME因子,或者在培养中缺乏TME因子。我们之前发表的研究表明,只有人类M2巨噬细胞(MØ)可以促进人类pdx衍生细胞的EMT,31我们测量了在MØ条件培养基(MCM)培养的pdx培养物中LOXL2的表达,并观察到明显的间充质形态转变(图2 b)以及细胞散射和运动增强(在线补充视频1和2),体外侵入(图2 c)及活体肝及肺定植(在线补充图S4A、B).在转录方面,MCM增加了几个EMT转录因子以及原型典型LOX和其他LOX样转录物的表达,其中EMT相关的LOXL2表现出最强的上调(图2 d),并在蛋白质水平得到证实(图2 e).

巨噬细胞通过OSM在pdx来源的PDAC细胞中诱导LOXL2和EMT。(A) LOXL2在指示的人PDXs中的表达。微管蛋白,加载控制。阳性(+)对照=过表达loxl2的293 T细胞裂解液。(B)用m2极化MØs (MCM)的对照培养基或条件培养基培养72小时的pdx衍生细胞的光镜照片。比例尺= 200µm。Insets = 2 x放大区域。(C)用对照培养基(Ctl)或MCM(左)培养72小时的Panc354 pdx衍生细胞的光镜照片。比例尺= 400µm。ctl处理或mcm处理的细胞通过0.8微米传输孔迁移的代表性图像(上,右),mcm处理的入侵细胞与ctl处理的Panc354、Panc215或Panc265细胞的平均折变±SD,设置为1.0。 (**P<0.01, ***p<0.001, unpaired Student’s t-test). (D) Mean fold-change ±SD of relative mRNA levels for the indicated genes in Ctl-treated or MCM-treated cells. Values normalised to ß-actin. Ctl-treated samples were set as 1.0. (*P<0.05, **p<0.01, ***p<0.001, unpaired Student’s t-test). (E) LOXL2 expression in Ctl-treated (–) or MCM-treated (+) PDX-derived cells. Tubulin, loading control. (F–H) OSM protein levels (pg/mL) present in (F) unpolarised PBMC-conditioned media or MCM, (G) unpolarised (Ctl) PBMC-conditioned media or conditioned media from TGFß1-treated PBMCs, or (H) serum from healthy controls or patients with PDAC. (*P<0.05, ***p<0.001, ****p<0.0001, unpaired two-sided Student’s t-test). EMT, epithelial to mesenchymal transition; LOXL2, lysyl oxidase-like protein 2; mRNA, messenger RNA; OSM, oncostatin M; PBMC, peripheral blood mononuclear cell; PDAC, pancreatic ductal adenocarcinoma; PDX, patient-derived xenografts; TGF, transforming growth factor.
我们之前发现MCM含有已知的EMT诱导剂TGFß1,31TGFß信号在LOXL2高表达的患者(即前四分位数)中持续上调(在线补充图S2B);然而,重组TGFß1 (rTGFß1)治疗不诱导EMT状态或LOXL2在人pdx衍生细胞中的表达(在线补充图S4C).因此,我们假设其他MCM EMT诱导因子可能介导LOXL2的表达。Oncostatin M (OSM)是一种合理的替代品,因为(1)垃圾等表明OSM是一种比TGFß1更有效的EMT诱导剂,尽管两者都汇聚成类似的pSTAT3/ smad3介导的EMT通路,38(2) OSM存在于MCM (图2 f(3) rTGFß1可诱导MØs分泌OSM (图2 g(4)与健康对照组相比,PDAC患者血清中OSM水平显著升高(图2 h).事实上,用重组OSM (rOSM)处理pdx来源的细胞可诱导间充质形态转变(在线补充图S4D)以及移民的增加(在线补充图S4E)和诱导emt相关基因的转录,包括LOXL2 (在线补充图S4C(下),表明MØ-secreted OSM可以诱导LOXL2的表达。我们还观察到MCM中高OSM浓度与pSTAT3激活和LOXL2蛋白表达之间的明确相关性(在线补充图S4F).重要的是,在Panc354细胞中沉默LOXL2并没有恢复rOSM或MCM在体外诱导emt样表型的能力(在线补充图S5A-C),表明LOXL2不是两种刺激诱导的emt相关表型的下游驱动因素。
的损失Loxl2改善OS,减轻肿瘤负担
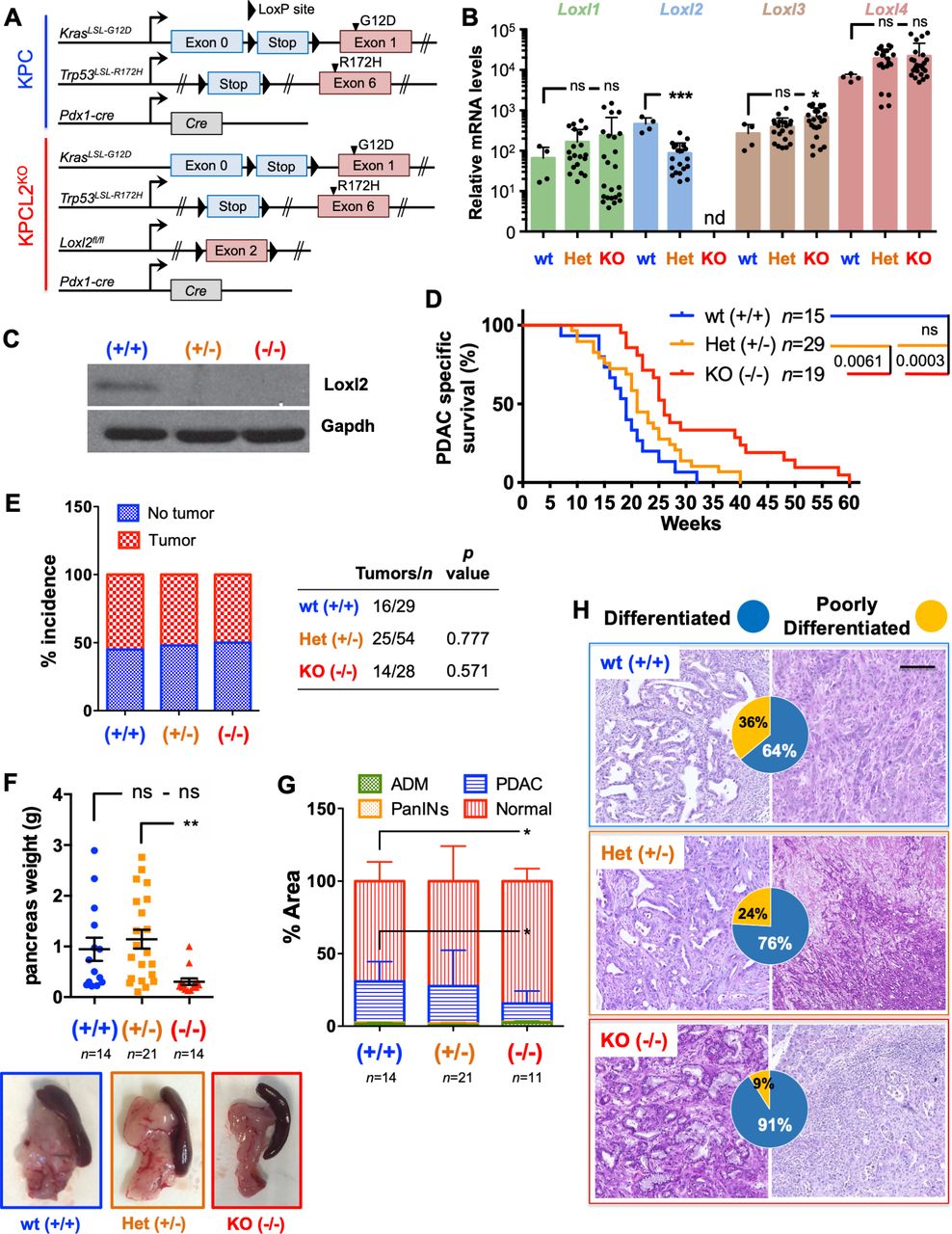
由于人类PDX模型不能概括人类TME (图2一个),我们进一步推断我们的发现到老鼠的系统。我们首先确认了四种主要小鼠PDAC KPC (Pdx1-cre;喀斯特LSL。G12D/+, Tp53LSL。R172H/+)衍生的细胞系表达无法检测的Loxl2水平,并且与人类PDAC肿瘤和细胞相似,小鼠MØ-secreted因子可以诱导Loxl2表达式(在线补充图S6A、B).具体来说,MCM或rTGFß1和rOSM在KPC细胞中诱导Loxl2和/或emt样状态(在线补充图S6B-F);然而,我们一致观察到,在小鼠设置中,rTGFß1是一个比rOSM更有效的EMT诱导剂。接下来,我们穿过floxedLoxl2等位基因的老鼠26用KPC小鼠生成KPC;Loxl2fl / fl敲除(KO)小鼠(称为KPCL2)KO)研究LOXL2在体内的作用(图3一).的损失Loxl2表达KPCL2KO肿瘤源性细胞或新鲜肿瘤分别通过定量反转录PCR (RT-qPCR)和western blot (WB)分析确认(图3 b, C).其他Lox-like mrna的水平不受蛋白质丢失的影响Loxl2,除了Loxl3(图3 b).接下来我们评估了KPC野生型(wt)、Het的OS、肿瘤发生率和肿瘤负担Loxl2(KPC;Loxl2+/-)和KPCL2KO小鼠,分为两个不同的队列(见方法)。在第一组中,损失Loxl2显著扩展的操作系统(图3 d),我们从第二个队列得出结论,这不是由于肿瘤发病率的差异(图3 e)而不是减少肿瘤负担(图3做减法而且在线补充图S7).有趣的是,Loxl2缺失也强烈影响肿瘤分化。然而KPC肿瘤是异质性的,64%的分析肿瘤表现出更高分化的表型,36%的肿瘤表现出低分化的表型,低分化肿瘤的百分比在KPCL2中下降到约9%KO老鼠(图3 h).

的损失Loxl2提高总体生存率,减少肿瘤负担。(A)胰腺癌基因小鼠模型方案。色码(蓝色,KPC野生型(wt);红色,KPCL2KO)用于所有结果。(B)来自指示基因型的肿瘤源性细胞中指示基因的平均相对mRNA水平±SEM (nd,未检测到;ns,不重要;* * * * p < 0.05, p < 0.001;单因素方差分析(ANOVA)与Dunnett’s后验)。(C) Loxl2在KPC基因型肿瘤匀浆中的表达。Gapdh加载控制。(D)存活率wt (+/+), Het (KPC;Loxl2+/-)和KO KPC (KPC;Loxl2-/-老鼠)。所有小鼠在指定时间死于PDAC相关疾病(p值显示;ns,不重要;log-rank (Mantel-Cox)测试)。计算中位生存期为:wt(+/+): 19周;Het (KPC;Loxl2+/-): 21周;和KO KPC (KPC;Loxl2-/-): 26周。(E) wt(+/+)中肿瘤发生率,Het (KPC;Loxl2+/-)和KO KPC (KPC;Loxl2-/-)小鼠出生后17-18周。(P值显示,偶然性分析,双面Fisher确切检验)。如果在尸检中肉眼可见肿瘤,则确定肿瘤发生率为阳性。(F)平均胰腺重量±SEM在wt (+/+), Het (KPC;Loxl2+/-)和KO KPC (KPC;Loxl2-/-)小鼠出生17-18周(左)。(* * P < 0.01;ns,不重要;单因素方差分析与Tukey后验)。出生后17-18周时PDAC肿瘤/基因型的代表性图像(下)。(G)从wt (+/+), Het (KPC;Loxl2+/-)和KO KPC (KPC;Loxl2-/-)小鼠出生后17-18周,分类为严重改变组织(腺泡到导管化生(ADM)和炎症),胰腺上皮内肿瘤(PanINs I-III),癌症组织(PDAC)或正常腺泡组织(*p<0.05,偶发分析,双侧Fisher确切检验)。(H)各肿瘤分级的代表性h&e染色切片:wt (+/+) KPC(蓝色,n=14), Het KPC (KPC;Loxl2+/-)(橙色,n=21)和KO KPC (KPC;Loxl2-/-)小鼠(红色,n=11)。饼图内嵌=每种基因型分化(蓝色)和低分化(黄色)肿瘤的百分比。比例尺= 250µm。Het,杂合的;KO,淘汰赛;Loxl2赖氨酸氧化酶样蛋白2;信使RNA信使RNA;PDAC,胰导管腺癌。
Loxl2过表达加重OS,增加肿瘤负担
接下来,我们将KPC小鼠与前面描述的条件小鼠过表达交叉Loxl226生成KPC;R26Loxl2-EGFPKI /吻敲入(KI)小鼠(称为KPCL2)KI) (图4一).EGFP和Loxl2在KPC衍生肿瘤(或细胞)中的过表达;R26+/+KPC;R26L2+ /吻KPC;R26L2KI /吻小鼠经荧光显微镜、WB和RT-qPCR分析证实图4 b, C)和过度表达Loxl2没有影响其他lox类成员的水平(图4 c).评估OS、肿瘤发生率和肿瘤负担,再次使用双队列方法,并与KPCL2对照KO小鼠的OS (图4 d)、肿瘤发病率增加(图4 e)和明显增加的肿瘤负担(图4 f-h在线补充图S7)时Loxl2过表达。这种肿瘤负担的增加与KPC中分化更差的表型相一致;R26L2+ /吻KPC;R26L2KI /吻小鼠,KPC为90%;R26L2KI /吻生长低分化肿瘤的小鼠(图4我).

过度的Loxl2恶化总体生存率,增加肿瘤负担。(一)基于kpc的方案Loxl2过表达基因小鼠模型。颜色代码为绿色,KPCL2KI,用于所有结果。(B)光和EGFP肿瘤图像(左)。Loxl2在KPC基因型肿瘤匀浆中的表达(右)。Gapdh加载控制。阳性(+)对照=小鼠过表达loxl2的293 T细胞裂解液。(C)指示基因型肿瘤源性细胞中指示基因的平均相对mRNA水平±SEM (***p<0.001;ns,不重要;单因素方差分析(ANOVA)与Dunnett’s后验)。(D) KPC野生型(+/+)存活率,KPC;R26L2+ /吻KPC;R26L2KI /吻老鼠。所有小鼠在指定时间死于PDAC相关疾病(p值显示;ns,不重要;log-rank (Mantel-Cox)测试)。计算中位生存期为:野生型(+/+):20周;KPC;R26L2+ /吻: 18周;KPC;R26L2KI /吻: 17周。(E) KPC野生型肿瘤发生率(+/+),KPC;R26L2+ /吻KPC;R26L2KI /吻出生后17-18周的小鼠。(* * * * P < 0.0001;ns,不重要;应变分析;双面Fisher确切检验)。如果在尸检中肉眼可见肿瘤,则确定肿瘤发生率为阳性。(F) KPC野生型平均胰腺重量±SEM (+/+), KPC;R26L2+ /吻KPC;R26L2KI /吻出生后17-18周的小鼠。* * (* P < 0.05, P < 0.01;ns,不重要;单因素方差分析与Tukey后验)。(G)出生后17-18周PDAC肿瘤/基因型的代表性图像。(H)野生型(+/+)KPC小鼠(蓝色,n=7)和KPCL2小鼠胰腺组织面积的量化KI(KPC;R26L2+ /吻KPC;R26L2KI /吻)小鼠(绿色,n=12)在出生后17-18周测定,分类为严重改变组织、PanINs I-III、PDAC或正常腺泡组织(*p<0.05, **p<0.01,偶然性分析,双侧Fisher确切检验)。(I)各肿瘤分级的代表性h&e染色切片:野生型(+/+)KPC(蓝色,n=6), KPC;R26L2+ /吻(深绿色,n=6)和KPC;R26L2KI /吻小鼠(浅绿色,n=6)。饼图内嵌=所示基因型的分化肿瘤(蓝色)和低分化肿瘤(黄色)的百分比。比例尺= 250µm。ADM, acinar-to-ductal化生;KI,敲入;Loxl2赖氨酸氧化酶样蛋白2;胰腺上皮内肿瘤;PDAC,胰导管腺癌;wt,野生型。
Loxl2在最初的PDAC开发中起作用
由于突变型p53通过基因组不稳定性加速肿瘤进展和恶性转化,导致突变增加,39 40我们分析了Loxl2wt中p53 KC过表达或缺失(喀斯特LSL。G12D/+;Pdx1-cre)小鼠发生腺泡导管化生(ADM)、胰腺上皮内肿瘤(PanIN)前体病变和PDAC,但其速度比KPC小鼠慢得多,因此可以评估最初的转化事件。与KPC小鼠相似,损失Loxl2的过表达增加了OSLoxl2有相反的效果(在线补充图S8A、B).关于胰腺重量,只有KC观察到显著增加;R26L2KI /吻老鼠(在线补充图S8C);然而,在组织学水平上,38-40周龄KCL2的胰腺KO小鼠主要由健康组织或有PanIN病变和ADM的组织组成,与wt小鼠相比,wt小鼠PDAC病变百分比更大且显著(p<0.001) (在线补充图S8DE).在KCL2KI与wt小鼠相比,PDAC的百分比显著增加(在线补充图S8D底,F)。
Loxl2丢失和过表达影响PDAC的转移和癌症干细胞特性
除了肿瘤负担和大小的差异外(图3和4)时,我们还观察到宏观转移的显著差异Loxl2调制(图5一个).转移发生率(宏观和组织学证实,在线补充图S9A、B) KPCL2KO和KPCL2KI将小鼠与它们各自的wt窝鼠进行比较,因为两个wt KPC菌株具有不同的背景,显示出基底转移的差异。符合生存,KPCL2KO小鼠的肝、肺和腹膜(肠、淋巴结、胃和/或横膈膜)转移灶较其他小鼠减少(图5 b).在KPCL2中观察到相反的情况KI小鼠转移率显著增高(图5 c),包括腹腔内转移,例如肠系膜淋巴结和膈肌转移(图5 a, C而且在线补充S9A B).在没有突变型p53的情况下,KCL2KO小鼠的转移发生率也显著降低,可能是这些小鼠PDAC病变发生率降低的直接结果,而在KC;R26L2+ /吻和KC;R26L2KI /吻小鼠的肝脏和肺转移的发生率增加(在线补充图S9C,D).

Loxl2丢失和过表达影响PDAC的转移和癌症干细胞特性。(A)来自特定基因型的PDAC肿瘤和转移(白色箭头,转移)。(B - c) (B)野生型(+/+),Het (KPC;Loxl2+/-)和KO KPC (KPC;Loxl2-/-)小鼠或(C) KPC野生型(+/+),KPC;R26L2+ /吻KPC;R26L2KI /吻出生后17-18周的小鼠。(* * P < 0.01, * * * * P < 0.0001;ns,不重要;偶发分析,双边费雪精确检验)。(D - e)左:肿瘤切片中(D)野生型(+/+)KPC和KO KPC (KPC;Loxl2-/-)小鼠或(E)野生型(+/+)KPC和KPCL2KI(KPC;R26L2KI /吻老鼠)。比例尺= 250µm。右图:阳性细胞百分比/感兴趣区域(ROI)的量化。(* * * * P < 0.0001;ns,不重要;未配对学生t)。(F)在指示细胞系(n=3个细胞系/基因型,****p<0.0001, ns,不显著,双面t检验+ Mann-Whitney U检验)播种后11天测定的平均数目%集落面积±SEM。(G)特定基因型肿瘤细胞系在播种后7天,平均球数(no.) /mL±SEM (*p<0.05, ***p<0.001,未配对Student 's t检验)。(H)指定基因型肿瘤细胞系中EpCAM+/CD133+细胞±SEM的平均百分比(***p<0.001,双面t检验+ Mann-Whitney U检验)。Het,杂合的; KI, knock-in; KO, knockout;Loxl2赖氨酸氧化酶样蛋白2;PDAC,胰导管腺癌;wt,野生型。
emt相关蛋白的表达与PDAC转移密切相关。41 42同样,我们之前已经证明了Loxl2以及EMT调控转录因子Snail1在一个乳腺癌小鼠模型中。23除了twist1阳性细胞的百分比增加外,在KPCL2中分析的其他emt相关蛋白的表达没有观察到差异KO肿瘤(图5 d而且在线补充图S10A).在KPCL2KI然而,小鼠所有蛋白质的表达均增加(图5 e而且在线补充图S10B),以配合增加的紧急医疗服务计划Loxl2是过表达。同样,正如预期的那样,在来自KPCL2的肿瘤细胞中检测到E-cadherin膜表达的减少KI与这些肿瘤低分化形态一致的小鼠(在线补充图S11A,B).有趣的是,当所有四种基因型的肿瘤衍生培养物用EMT诱引物TGFß1或Osm处理时,所有细胞在形态、转录和蛋白质水平上对rTGFß1或rosm刺激都有反应(尽管有差异)(在线补充图S12和13),表示无论存在或不存在Loxl2,这些细胞是tgf ß1响应和osm响应和emt胜任。
我们还评估了影响肿瘤负担和转移的癌症干细胞(CSCs)的百分比41在KPCL2KO和KPCL2KI文化。Loxl2过表达显著增加集落形成能力(图5 f),这可能解释了在KPCL2中观察到的肿瘤重量和大小的增加KI老鼠。同样,在KPCL2中,球的形成能力(即自我更新)和CSCs的比例(测定为EpCAM+/CD133+)均显著增加KI但在KPCL2中减少KO细胞(图5 g, H),这表明Loxl2表达式可能链接到CSC状态。尽管如此,在极限稀释试验中,KPCL2的肿瘤频率没有差异KO或KPCL2KI细胞与它们各自的wt对照(在线补充图S14A,B),注射KPCL2后,观察到肿瘤大小和重量有显著差异KO细胞(在线补充图S14A),提示肿瘤起始和/或生长动力学降低。
Loxl2是PDAC细胞内穿刺的必要条件吗
KPCL2的容量KO和KPCL2KI测定小鼠尾静脉注射后肺定植细胞的实验转移能力。既不存在也不存在Loxl2影响KPCL2的转移能力KO和KPCL2KI在体内形成肺转移的细胞(图6 a, B),这表明Loxl2表达不影响注入循环系统的pdac源性细胞的实验转移能力(即外渗、播撒和定植),而是可能通过非细胞自主机制阻碍或促进原发肿瘤细胞的侵袭和/或内渗。因此,我们首先证实了Loxl2是通过KPCL2分泌的KI而不是KPCL2KO细胞(在线补充图S14C),然后通过注射h2b - mcherry标记的KPCL2在体内建立原位肿瘤KO或EGFP + KPCL2KI细胞,单个或按1:1比例组合(图6 c).如果KPCL2KO由于缺乏胞外Loxl2,细胞内渗透能力下降,外源性Loxl2由KPCL2表达KI细胞应该挽救这种表现型。注射4周后,所有小鼠体内都形成了重量相等的肿瘤(图6 d),与预期的mcherry阳性和egfp阳性细胞分布(图6 e).正如假设的那样,h2b - mcherry标记的KPCL2KO与EGFP+KPCL2相比,循环中的细胞数量明显减少KI细胞,(图6 f);然而,当EGFP+KPCL2以1:1的比例建立肿瘤时,小鼠血液和肝脏中均检测到mcherry阳性细胞的显著增加KI细胞(图6 f, G),证实了KPCL2KO当提供外源性Loxl2时,细胞能够在肝脏内渗透并播撒种子。
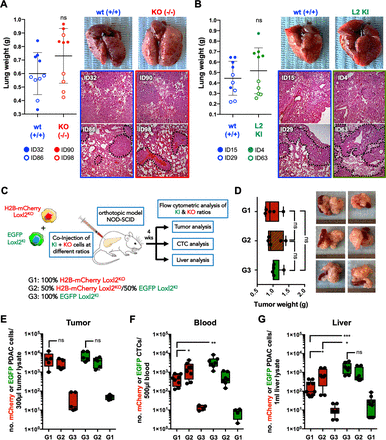
Loxl2是PDAC内穿刺的必要条件。(A - b)野生型(+/+,ID32) KPC和Het KPC (KPC;Loxl2+/-, ID86)细胞(蓝色)和KO KPC (KPC;Loxl2-/-(B)野生型(+/+,ID15和ID29) KPC细胞(蓝色)和KPCL2KI(KPC;R26L2+ /吻, ID63和KPC;R26L2KI /吻, ID4)细胞(绿色)在NOD-SCID免疫缺陷小鼠(左)注射后4周(ns,不显著;未配对学生t)。提取肺的代表性图像(上,右)和H&E图像(下,右)。(C)用h2b - mcherry标记的KPCL2在NOD-SCID免疫缺陷小鼠体内原位肿瘤建立的实验装置KO(KPC;Loxl2-/-)细胞或EGFP+KPCL2KI(KPC;R26L2KI /吻)细胞,单独或按1:1比例(分别为1、3、2组)。(D)原位注射后4周,每组(n=5只)平均肿瘤重量±SD(左)。PDAC肿瘤的代表性图像(右)。(E - G)各组(E)肿瘤匀浆、(F)血液或(G)肝脏匀浆中mcherry阳性(红色)或egfp阳性(绿色)细胞的平均数量(个数)(*p<0.05, **p<0.01, ***p<0.001;ns,不重要;未配对学生t)。Het,杂合的;KI,敲入;KO,淘汰赛;Loxl2赖氨酸氧化酶样蛋白2;PDAC,胰导管腺癌;wt,野生型。
Loxl2影响胶原纤维定向和转移前小生境形成
LOX和LOX样蛋白在不同肿瘤系统中的转移促进作用与细胞外功能有关,如ECM组织,特别是胶原纤维交联导致组织僵硬19 43].事实上,具有高上皮张力的硬性肿瘤与更具侵袭性的PDAC表型相关,导致患者生存时间较短,10原发肿瘤ECM组织可以增强因子(如外泌体)的分泌,通过ECM重塑或基质细胞募集,促进继发器官的转移前生态位调节,最终促进转移。
因此,我们首先分析了肝脏中骨髓来源的MØ群体,该群体已被证明在PDAC细胞转移中至关重要,44 45作为观察到的KPCL2转移差异的潜在解释KO和KPCL2KI老鼠。分析结果显示CD45的存在呈负相关和正相关+CD11b+F4/80+细胞46在KPCL2KO和KPCL2KI小鼠,分别为(图7).我们还评估了PDAC肿瘤的ECM,特别是胶原纤维的方向性/组织,这与Loxl2和肿瘤细胞直接相关。47而两种KPCL2的方向性直方图wt和KPCL2KI肿瘤在特定/偏好方向表现出单一的显性峰,KPCL2KO肿瘤包含多个峰,表明完全各向同性行为(图7 binsets)。相对峰值频率的定量证实,在缺乏的情况下,ECM组织明显较差Loxl2当过度表达时,组织性增强(图7 c).根据胶原纤维排列的差异,我们观察到KPCL2中pSTAT3和pFAK显著增加KIpSTAT3和KPCL2中pFAK表达减少的趋势KO肿瘤(在线补充图S15A,B),表明上皮张力和硬度的差异Loxl2是调制。10 14 48我们还通过pMLC-2染色评估了机械收缩力,48在KPCL2中观察到活性pMLC-2的较高表达KI与KPCL2KO肿瘤(在线补充图S15A,B).最后,为了对KPCL2中的免疫TME有一个更全面的了解KO和KPCL2KI小鼠进行免疫组化(IHC)和流式细胞术分析(在线补充图S16).当比较KPCL2时,我们确实观察到免疫时间me的差异KI和KPCL2KO肿瘤与它们各自的窝伴侣对照。有趣的是,我们观察到KPCL2中CD3染色增加KO和KPCL2KI肿瘤(在线补充图S16A,B),可通过流式细胞术进行验证(在线补充图S16C,D).同样,KPCL2中巨噬细胞数量(如F4/80+),特别是M2亚群(如CD206+)增加KI肿瘤和KPCL2减少KO模型,与它们各自的小窝伴侣对照进行比较,并根据所使用的方法(在线补充图S16A-D).最后,虽然CD45免疫组化染色在福尔马林固定、石蜡包埋(FFPE)样品中不稳定,但流式细胞术分析了KPCL2中的CD45KO和KPCL2KI与相应的wt对照相比,肿瘤分别表现出显著的减少和增加。由此可见,缺乏或过度表达Loxl2会影响TME免疫细胞形态;然而,在得出任何明确的结论之前,还需要对具有相似基因背景的小鼠进行更详尽的分析。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Loxl2影响胶原纤维定向和转移前小生境形成。(A) (top)野生型(+/+)KPC和Het KPC (KPC;Loxl2+/-)老鼠(蓝色)和KO KPC (KPC;Loxl2-/-)小鼠(红色)或(下)野生型(+/+)KPC小鼠(蓝色)和KPCL2KI(KPC;R26L2+ /吻KPC;R26L2KI /吻)小鼠(绿色)在出生后14-16周测定。(* P < 0.05, * * * * * P < 0.01, P < 0.001;ns,不重要;双面t检验+ Mann-Whitney U检验)。(B)胰脏代表性胰液红染色图像。覆盖,有代表性的纤维方向性分析图,描述纤维在特定方向的频率。(C)如(B)所示胰脏苦葡萄糖红染色图像的方向分析图的平均最大相对(最大rel.)峰值频率±SEM。(n=每个基因型5-6只小鼠,每只小鼠分析4-6张代表性图像,*p<0.05, ****p<0.0001,双面t检验+ Mann-Whitney U检验)。(D)氯膦酸盐(Clod)体内研究的实验装置。(E)平均折变Loxl2(上)或Osm(下)对照组脂质体处理与氯膦酸盐脂质体处理的KPC wt小鼠的胰腺相对mRNA水平±SEM。价值观被正常化产生Hprt水平和对照脂质体处理的样品设置为1.0。(n=每组8只小鼠,****p<0.0001,双面t检验+ Mann-Whitney U检验)。(F)对照(Ctl)和氯膦酸盐处理的KPC wt小鼠胰腺代表性的苦葡萄糖红染色图像和覆盖纤维方向性分析图。(G)胰脏苦参红染色图像的方向分析图的平均最大相对(最大rel.)峰值频率±SEM见(F)。(n=每组5只小鼠,每只小鼠分析3张代表性图像,***p<0.001,双侧t检验+ Mann-Whitney U检验)。(H)在指定组中检测到的肿瘤和转移的数量。Het,杂合的;产生Hprt,次黄嘌呤磷酸核糖转移酶1;KI,敲入;KO,淘汰赛;Loxl2赖氨酸氧化酶样蛋白2;信使RNA信使RNA;Osm制瘤素M;PDAC,胰导管腺癌;wt,野生型。
最后,使用同源原位PDAC异种移植模型,小鼠分别用对照或氯膦酸酯脂质体消除MØs (图7 d),正如我们此前报道的那样。49事实上,大幅减少Osm而且Loxl2表达式(图7 e)在氯膦酸钠治疗小鼠的PDAC肿瘤中测定,伴随胶原纤维定向的显著下降(图7 f),相对峰值频率降低(图7 g)和减少整体转移(图7 h),确认MØs生产Osm,诱导Loxl2并促进ECM的正常组织,这是转移所必需的。
讨论
在此,我们将重点放在LOXL2上,因为之前发表的研究表明,LOXL2在PDAC样本或细胞系中过表达和分泌,50 51其表达与PDAC转移和化疗耐药呈正相关。33 52-54然而,这些研究中的大多数要么是观察性的,要么是建立在PDAC细胞系中LOXL2小干扰RNA沉默的基础上的。同样,迄今为止,没有研究从遗传学上检验LOXL2在PDAC免疫小鼠模型中的作用,该模型概括了人类疾病的所有方面。
我们观察到人类PDAC PDXs和pdx衍生细胞系表达低到无法检测到的Loxl2蛋白水平,这强调了使用小鼠模型来准确研究Loxl2在PDAC中的作用。在已建立的PDAC细胞系中,其他研究也观察到类似的结果,各种研究一致认为,只有间充质样PDAC细胞(如MiaPaCa和Panc1)表达LOXL2。33 54沿着这些线索,只有Panc265,一种最近被证明在体内高度转移的PDX,37LOXL2表示。因此,我们生成了缺乏和过表达的具有免疫能力的KPC和KC小鼠Loxl2准确的研究Loxl2在活体环境中。Loxl2缺失和过表达最显著的结果分别是抑制和增强转移,这转化为KCL2 OS的增加和减少KO/ KPCL2KO和KCL2KI/ KPCL2KI老鼠,分别。我们还观察到KPCL2基因型在体内瘤内异质性的明显差异KI小鼠产生更多低分化肿瘤,EMT因子表达增加。后者与观察到的情况有关LOXL2在鳞状PDAC肿瘤中表达更高,其特征是侵袭性增加,EMT和OS降低。我们之前也证实了多瘤病毒中T抗原(PyMT)肿瘤中Loxl2/去分化之间的关系,23并描述了Loxl2对EMT的积极调节作用,特别是对Snail1的稳定性和功能活性的积极调节作用。23日25此外,在KPCL2中观察到正相关KI干细胞水平的细胞。因此,EMT和stemness的增加可以解释在KPCL2中观察到的转移增强KI老鼠。
有趣的是,KPCL2KOwt肿瘤表达相似水平的EMT相关蛋白,对EMT诱导剂TGFß1或Osm的反应相似。同样,KPCL2KO细胞能够定植并形成肺大转移到与wt KPC和KPCL2相似的水平KI这表明细胞自主的Loxl2并不影响这些细胞的emt反应性或转移能力。然而,由于浸润和静脉内注射先于转移性定植和生长,丧失Loxl2可能影响转移级联的这些关键早期事件,这也依赖于非细胞自主因子和过程,如TME基质硬度,它可以激活肿瘤细胞EMT,侵袭和转移。10 55另一方面,根据KCL2的数据KO在小鼠和ELDA致瘤性实验中,Loxl2也可能在肿瘤起始中起重要作用。尽管如此,由于LOXL2的功能之一被描述得最好的是胶原纤维的交联、稳定和组织,16我们推断在KPCL2中观察到转移减少KO小鼠的发病主要(但不完全)由于胞外Loxl2介导的外源性因素。事实上,我们能够挽救KPCL2的体内转移能力KO通过共注射KPCL2提供细胞外Loxl2来抑制肿瘤来源细胞KI细胞。同样,KPCL2中的胶原纤维方向性和组织,以及机械信号相关蛋白(pSTAT3, pFAK和pMLC-2)KO和KPCL2KI原发肿瘤表现出相反的表型,但在ECM的数量上没有观察到明显的差异,证实了KPCL2中基质组织(和硬度)受到损害KO肿瘤。这与Collison及其同事最近报道的数据不同,在那里作者表明(1)基质基质抑制而不是促进PDAC,(2)当使用抗loxl2单抗(AB0023, Gilead)消除小鼠同基因PDAC模型中的ECM时,小鼠PDAC进展增强。14作者将其归因于ECM含量和纤维化的减少。最近,Kalluri和同事们还发现,体内肌成纤维细胞衍生的Col1缺失会加速PanINs和PDAC的出现。15
在我们的研究中,KPCL2KO小鼠OS改善,转移减少,而KPCL2则相反KI老鼠。重要的是要注意在KPCL2中KO老鼠,Loxl2仅在PDAC细胞和出生时基因缺失,因此与江等研究14不能做。同样,我们不能排除TME中存在的其他细胞类型可能代表Loxl2的替代来源;然而,这种潜在的贡献可能是最小的,因为胶原纤维的交联和组织在KPCL2中一直受到损害KO肿瘤。在我们的同生PDAC体内系统中,而胶原纤维错位而减少Loxl2是通过氯膦酸盐治疗实现的,类似于,14PDAC转移没有增加,而是减少。后者可能是氯膦酸酯脂质体介导的额外巨噬细胞耗竭的结果,其他人已经证明这是PDAC转移所必需的。56事实上,我们观察到免疫TME受Loxl2水平,这可能有助于观察到的体内表型独立或与ECM组织结合。最后,2017年simtuzumab(一种人源化IgG4单克隆抗体(源自AB0023),可抑制细胞外LOXL2)与吉西他滨联合用于转移性PDAC患者一线治疗的随机II期研究的结果显示(ClinicalTrials.gov),NCT01472198),表明在吉西他滨的基础上加入simtuzumab并不能改善临床结果57;然而,联合治疗并没有加速肿瘤的进展。因此,这些关于LOXL2在PDAC成瘤过程中作用的不同观点突出了肿瘤-基质相互作用的复杂相互作用,需要进一步了解这种相互作用,并重新评估抗LOXL2抗体或小分子LOXL2抑制剂的效用58在转移性PDAC的治疗中。
在本研究中,我们还发现巨噬细胞和OSM是这种相互作用的重要参与者。有趣的是,Dinca等最近在浸润性乳腺导管癌中发现,OSM可诱导LOXL2表达,进而促进ECM I型胶原纤维交联增加,导致细胞侵袭性增加。59同样,李等,最近证实MØ-secreted Osm在CAFs中诱导炎症基因表达,在PDAC中创造一个促肿瘤发生的环境。60有趣的是,他们还显示肿瘤细胞被植入Osm−−/小鼠表现为上皮主导型形态,肿瘤生长和转移减少。因此,我们的研究结果不仅明确地将LOXL2的表达与PDAC中巨噬细胞分泌的OSM联系起来,而且综合起来,这些研究强调了靶向巨噬细胞、OSM或两者(例如,使用抑制剂靶向CSF1R或GP130)可能为治疗PDAC提供了一种更有效的治疗策略,并规避了单独靶向LOXL2的潜在问题。
数据可用性声明
本文中使用的数据集的数据可在一个公共的、开放的存储库中获得。其他资料应合理要求提供。https://www.nature.com/articles/ng.3398;https://bmccancer.biomedcentral.com/articles/10.1186/s12885-016-2540-6;//www.marcconsult.com/content/66/9/1665。
伦理语句
病人同意发表
伦理批准
根据当地Salud Carlos III研究所动物实验伦理委员会(PA 34-2012)或Autónoma de Madrid (UAM)大学动物护理使用委员会(Ref# CEI-25-587)和马德里大学(PROEX 335/14, 182/14或294/19)批准的协议,在小鼠身上进行人体PDXs的活体扩展、小鼠繁殖和所有体内程序。在所有体内实验中,根据机构准则安置小鼠,所有实验程序都按照实验动物福利的机构准则和根据国际医学科学组织理事会(医学科学组织理事会)制定的《涉及动物的生物医学研究国际指导原则》中所述的《照料和使用动物的道德行为准则》进行。来自PDAC患者和健康献血者的血液样本由生物银行医院Ramón y Cajal-IRYCIS (PT13/0010/0002)提供,该医院纳入了西班牙国家生物银行网络(ISCIII生物银行注册号为;B.0000678)和ACar来自卡洛斯三世研究所(ISCIII ref n°:C.0003953)的“家族性胰腺癌登记处样本收集”。样品按照标准操作程序进行处理,并得到伦理和科学委员会(控制编号:不。对照:DE-BIOB-73 AC65, RG。B我OB-57 and RG.BIOB-54), with informed consent and according to Declaration of Helsinki principles. Tumours derived from surgical resections at Rechts der Isar Hospital, Technical University of Munich, were obtained with written informed consent and authorised by the Rechts der Isar Hospital Ethics Committee with Project number 1926/07 and 5510/12.
致谢
我们感谢患者和与西班牙国家生物银行网络整合的生物银行医院Ramón y Cajal-IRYCIS (PT13/0010/0002)的合作,特别是Adrián Povo Retana的巨噬细胞分离和Javier Fco博士。雷加德拉·冈萨雷斯,组织学协助。我们还要感谢Vanesa Bermeo、Emilio González-Arnay和Sandra Batres Ramos为这项研究提供的宝贵技术帮助。SV和PS-T获得了西班牙马德里的马德里大学奖学金,Ayudas Para La Contratación de Predoctorales y Técnicos de Laboratorio(分别为PEJD-2017-PRE/BMD-5062和PEJ-2017-TL/BMD-7505)。
参考文献
脚注
ACan和BS,J是联合资深作者。
推特@kivancgorgulu, @sainz_lab
MA-N和LR-C贡献相同。
贡献者MV发起了这项研究。MA-N、PS、SA、KG、MV、MO、PG-S、JCL-G、SV和LM-H进行体外实验并分析数据;MV、CP、LY和LR-C进行体内研究并分析数据;LG-B进行RNA FFPE提取;PM、KG和SMW进行生物信息学分析;KG、PS-T、CS-P、DK进行组织学分析;EGG是我们的内部病理学家,并协助PCH和MO进行体内组织评估;GM-B对人类PDAC肿瘤进行免疫组化分析;CH、PS和SMDT领导了OSM研究;JE和ACar进行ELISA分析,并提供患者原血清和肿瘤样本; ME provided human resected tumor samples for RNA analyses; AM, FS, ACan and FP produced and characterised the Loxl2 mouse models. HA, KG, PCH and GM-B provided significant scientific input, analysed data and reviewed the manuscript. ACan, FP and BSJ developed the study concept, obtained funding, interpreted the data and drafted/edited the manuscript. BSJ acts as the guarantor, and accepts full responsibility for the finished work and/or the conduct of the study, had access to the data, and controlled the decision to publish. All authors edited the manuscript.
资金JCL-G获得了“la Caixa”基金会(ID 100010434)奖学金(LCF/BQ/DR21/11880011)的支持。本研究得到ISCIII FIS资助PI18/00757和PI21/01110 (BSJ)和PI18/00267 (lgb),以及西班牙经济和创新部SAF2016-76504-R (ACan和FP)、pid2019 - 111052rm - i00 (FP)、pid2019 - 104644rm - i00 (GM-B)、Ramón y Cajal Merit Award RYC-2012-12104 (BSJ)和ISCIII、CIBERONC、CB16/12/00446 (ACar)和CB16/12/00295 (ACan和GM-B)的资助,所有这些研究都是通过Fondo Europeo de Desarrollo Regional (FEDER)共同资助的。“Una manera de hacer Europa”;Fero基金会基金(BSJ);来自Fundación Científica Asociación Española Contra el Cáncer (FC-AECC) (BSJ)的协调拨款(GC16173694BARB);米格尔·塞雷特奖(CP16/00121) (PS);a德国研究基金资助项目:492 436 553 (KG);和德国癌症援助的Max Eder奖学金(111746)(PCH)
相互竞争的利益没有宣布。
患者和公众的参与患者和/或公众未参与本研究的设计、实施、报告或传播计划。
来源和同行评审不是委托;外部同行评议。
补充材料本内容由作者提供。它没有经过BMJ出版集团有限公司(BMJ)的审查,也可能没有经过同行评审。讨论的任何意见或建议仅仅是那些作者(s)和不被BMJ认可。BMJ放弃从放在内容上的任何依赖产生的所有责任和责任。如果内容包含任何翻译材料,BMJ不保证翻译的准确性和可靠性(包括但不限于当地法规、临床指南、术语、药品名称和药物剂量),并且不对翻译和改编或其他原因引起的任何错误和/或遗漏负责。
请求的权限
如果您希望重用这篇文章的任何部分或全部,请使用下面的链接,它将带您访问版权清除中心的RightsLink服务。您将能够快速获得价格和以多种不同方式重用内容的即时许可。