


摘要
硫嘌呤类药物用于治疗肿瘤和自身免疫性疾病患者以及移植受者。这些物质在某种程度上是由年代-甲基化由硫嘌呤甲基转移酶(TPMT)催化。近20年前,人类组织中TPMT活性水平由一种常见的遗传多态性控制的发现,成为药物遗传学对临床医学潜在重要性的最佳例子之一。具体来说,现在已知的是,遗传性TPMT活性水平非常低的患者在使用这些药物的标准剂量治疗时,发生硫嘌呤诱导的毒性(如骨髓抑制)的风险大大增加,而活性非常高的受试者可能治疗不足。此外,最近的报道表明,TPMT可能是临床重要药物相互作用的靶点,这种常见的遗传多态性可能是治疗依赖性继发性白血病发生的危险因素。在这些临床报告的同时,通过克隆和鉴定人类TPMT cDNA和基因,确定了TPMT多态性的分子基础。这些进展导致了一系列单核苷酸多态性的描述和表征,这些多态性导致低水平的酶活性,以及可能“调节”酶活性水平的TPMT基因5 ' -侧翼区域内的多态性变量数串联重复。由于这些观察结果,TPMT遗传多态性代表了基本药物遗传学信息开发和应用于临床医学的模式系统。
6-巯基嘌呤、6-硫代鸟嘌呤和硫唑嘌呤是巯基嘌呤类药物,用于治疗急性淋巴细胞白血病、自身免疫性疾病、炎症性肠病和器官移植受者(帕特森和蒂德,1975年;林纳德1992).硫嘌呤类药物是非常有用的药物,但它们的治疗指数相对较窄,主要毒性是危及生命的骨髓抑制(帕特森和蒂德,1975年;林纳德1992).这些药物在某种程度上是由年代-由细胞质催化的甲基化年代-adenosyl -l-蛋氨酸依赖酶硫嘌呤甲基转移酶1电子商务2.1.1.67) (雷米,1963;Woodson和Weinshilboum, 1983).人体组织中TPMT的活性水平是由一种常见的遗传多态性(Weinshilboum和Sladek, 1980年),而这种多态性提供了药物遗传学对临床医学潜在影响的一个较好的例子(Weinshilboum等人,1999年).因此,TPMT多态性是一个模型系统,它说明了药物遗传学信息转移到临床的过程,以及过去20年药物遗传学研究从表型研究迁移到包括基因型研究的过程。随后的讨论将简要回顾如何将对TPMT的生化药物遗传学理解“转化”为临床实验室为基础的检测,这有助于使个性化硫嘌呤治疗成为可能。此外,平行过程的分子理解的TPMT多态性已发生将被描述。最后,关于这一药物遗传学研究领域的未解问题也将进行总结。
TPMT:临床研究
年代-甲基化被报道为硫嘌呤药物最初用于人体后不久的代谢途径(Elion 1967).Remy在20世纪60年代早期使用啮齿动物组织(雷米,1963).然而,直到20世纪70年代末,TPMT的活性才首次在人体组织中进行分析和研究(Weinshilboum等人,1978),其明确的目标是测试这一假设,即硫嘌呤生物转化途径的个体变异可能与药物毒性和/或治疗效果的个体差异有关。这些最初的人体TPMT酶测定是用一种容易接近的组织——红细胞(RBC)进行的。这样做是为了开发一种临床测试,如果可以证明RBC TPMT活性与硫嘌呤代谢部位(如肝脏)的酶活性相关。
红细胞硫代嘌呤甲基转移酶活性测定的首次应用涉及在大群体样本和核心家族中进行的药物遗传学实验(Weinshilboum和Sladek, 1980年).这些研究表明,红细胞TPMT活性水平的“性状”是由一种常见的遗传多态性控制的。所研究的白种人中,大约89%的人具有一个或多个导致高活性的基因纯合子;11%为杂合子,具有中等活性;每300个受试者中有1个是纯合子,具有RBC TPMT活性低的特征(图2)。1).回想起来,幸运的是,最初的TPMT药物遗传学研究是在白种人受试者中进行的。如果在东亚进行,如随后所述,三模态频率分布如图所示。1不会被观察到(Jang等人,1996;Park-Hah等人,1996).在1980年首次以种群和家族为基础的RBC TPMT药物遗传学研究被报道后,在这十年的剩余时间里,关于多态性的工作集中在旨在表征酶的生化特性的实验上(Woodson和Weinshilboum, 1983;伍德森等人,1983年;Ames等人,1986年),以及实验证明,由基因决定的红细胞中TPMT活性水平反映了其他人体组织和细胞(如肾脏、肝脏和淋巴细胞)中TPMT酶活性和免疫反应蛋白的相对水平(Szumlanski等人,1992年;Van Loon和Weinshilboum, 1982;伍德森等人,1982年).

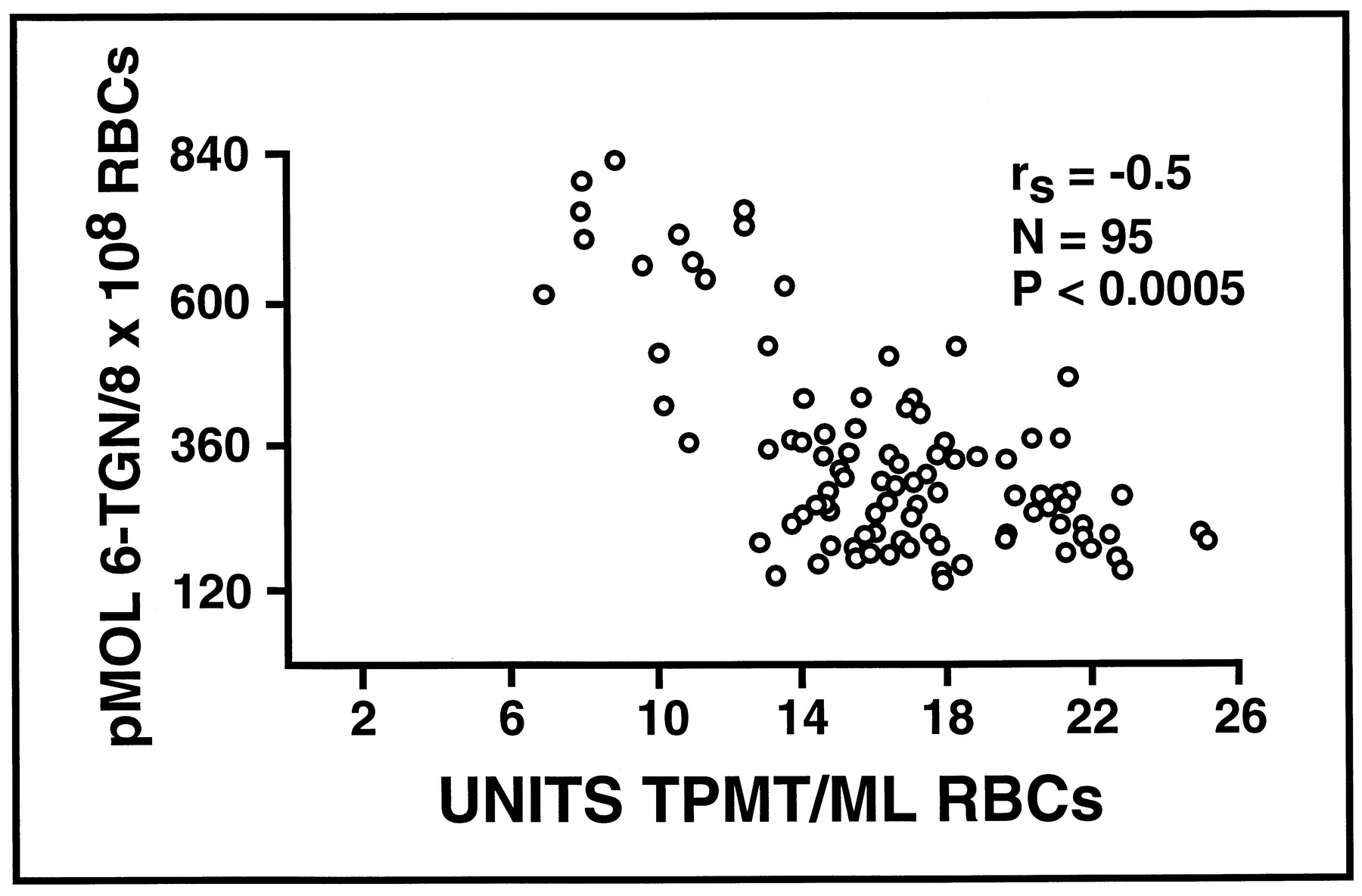
20世纪80年代,人类组织中TPMT的生物化学和调控知识稳步增长,与此同时,人们对巯基嘌呤药物在人体中的生物转化的了解也在不断增加。研究表明,6-巯基嘌呤(6-巯基嘌呤)等巯基嘌呤是前药物,可通过代谢激活形成6-硫鸟嘌呤核苷酸(6-TGNs) (伦纳德和马多克斯,1983年).红细胞中测定的6-TGNs水平与硫嘌呤治疗效果和毒性(如骨髓抑制)相关(Lennard等人,1983年).然而,为什么用相似或相同剂量的硫嘌呤药物治疗的患者RBC 6-TGN浓度可能有很大差异的问题仍然没有答案。巯基嘌呤临床药理学与硫代嘌呤药物遗传学研究的结合导致人们认识到,尽管巯基嘌呤是几种代谢途径的底物(见图2)。2),导致6-TGN浓度个体差异的主要因素是由基因决定的TPMT活性水平(Lennard等人,1987年,1990).具体而言,TPMT活性水平越高,RBC中6-TGN水平越低,反之亦然(图2)。3.).从这些观察到1989年的第一份报告仅一步之遥,即基因上TPMT活性非常低或不存在的患者,如果使用这些药物的“标准”剂量治疗,则有发展危及生命的硫嘌呤毒性的风险(Lennard等人,1989年).此外,有证据表明,TPMT活性非常高的患者在使用标准剂量的硫嘌呤药物治疗时,其疗效可能会下降(Lennard等人,1990年).这些相同的临床研究也提出了在慢性药物治疗期间诱导TPMT活性水平的可能性。具体而言,观察到在白血病药物治疗终止后,红细胞酶活性水平平均下降约25% (Lennard等人,1990年),表明在慢性治疗过程中酶活性增加。这个问题将在下面再次讨论硫代嘌呤甲基转移酶:悬而未决的问题.总之,这些发展提供了一个增强的能力个性化硫嘌呤药物治疗的前景。从那些早期的临床报告开始,极低的TPMT活性与硫嘌呤毒性的关联已被反复证实(Weinshilboum等人,1999年).因此,在一些转诊中心,TPMT酶活性的测量已成为标准的临床试验(梅奥基金会实验室医学公报,1991年),这是药物遗传测试进入临床实践主流的首批例子之一。然而,将这一药物遗传学信息转化为医疗实践所需的时间应该得到注意。1989年首次观察到基因上低TPMT活性与药物毒性风险增加有关(Lennard等人,1989年),随后在20世纪90年代出现的一系列临床报告中得到了证实。这些报告涉及各种疾病,如急性淋巴细胞白血病、皮肤病、肾和心脏移植、类风湿关节炎和自身免疫性肝炎(埃文斯等人,1991;Anstey等人,1992年;Schütz等,1993;Kerstens等人,1995年;阿里等人,1995年;Escousse等人,1995年).这些临床报告的数量在20世纪90年代逐渐增加,在20世纪90年代中期达到顶峰。因此,在首次报告遗传性TPMT活性降低与威胁生命的硫嘌呤诱导的骨髓抑制之间的联系后,临床验证和确认过程需要近十年的时间。将这一时间过程与未来药物遗传学信息的翻译过程进行比较将是有趣的,届时基于dna的技术的进步可能使以更快速和更具成本效益的方式为临床医生提供药物遗传学数据成为可能。

Thiopurine生物转化。
硫唑嘌呤和6-巯基嘌呤生物转化生成6-TGN的简化概述。XO,黄嘌呤氧化酶;次黄嘌呤磷酸核糖转移酶;IMP,肌苷单磷酸;GMP,鸟苷单磷酸。

急性淋巴母细胞白血病(ALL) 95例急性淋巴母细胞白血病(ALL)患儿RBC TPMT活性与RBC 6-TGN浓度的关系
修改后的Lennard et al. (1990).转载须经《柳叶刀》.
了解TPMT在巯基嘌呤生物转化中的作用提供了超越药物遗传学的见解,包括临床重要药物相互作用的可能性。在20世纪80年代初对TPMT进行生化表征期间,人们注意到苯甲酸衍生物如水杨酸是该酶的有效抑制剂(伍德森等人,1983年;Ames等人,1986年).这些观察结果提高了药物相互作用的可能性,如果TPMT被证明在硫嘌呤毒性和/或治疗效果的个体差异中发挥重要作用。然而,它需要十多年的第一个报告,可能的相互作用之间的硫嘌呤和TPMT抑制剂,如用于治疗炎症性肠病的氨基水杨酸衍生物(格里芬和迈纳,1995年;Hanauer 1996).磺胺柳氮和其他氨基水杨酸衍生物用于治疗这些患者被证明是有效的重组人TPMT的体外抑制剂(Szumlanski和Weinshilboum, 1995),目前至少有一份报告显示,当这些药物被施用于一名同时接受标准剂量硫嘌呤药物治疗的患者时,可能会发生严重的药物相互作用(刘易斯等人,1997年).该报告提出了这样的可能性,即这些患者可以转化为基因上TPMT活性低的受试者的“表型复制”。
最后,TPMT多态性的临床意义可能不会以急性毒性、治疗效果的变化或药物相互作用而结束。一段时间以来,人们已经知道使用抗肿瘤药物治疗可能与晚期继发性瘤变的发生有关,这被认为是最初的化疗造成的。例如,患有急性淋巴细胞白血病的儿童,在接受包括巯基嘌呤在内的药物治疗方案后,最初被“治愈”,后来可能发展为急性骨髓性白血病,这被认为与治疗有关(Ratain和Rowley, 1992年).虽然许多因素都涉及到这种药物治疗的悲剧性结局的发生,但目前已有一些报告表明,TPMT活性下降可能是这些患者发生继发性骨髓增生异常综合征或急性骨髓性白血病的危险因素之一(Relling等人,1998年;Thomsen等人,1999年).
综上所述,TPMT遗传多态性是药物遗传学在临床应用的一个很好的例子。基因上TPMT活性低的受试者发生硫嘌呤诱导毒性的风险大大增加,而TPMT活性非常高的受试者可能需要稍微增加这些药物的剂量进行治疗——始终要进行仔细的临床监测。此外,当患者同时使用巯基嘌呤药物和已知抑制TPMT的氨基水杨酸衍生物等药物治疗时,应谨慎。最后,TPMT活性降低可能是使用硫嘌呤治疗的患者发生晚期继发性瘤变的危险因素,这一可能性值得进一步研究。与此同时,与此相辅相成的是,对TPMT遗传多态性的临床相关性的快速发展的理解是对TPMT遗传调控的分子基础知识的稳步增长。
TPMT:分子研究
了解药物反应遗传变异的分子基础一直是药物遗传学研究的主要目标。因此,在前文所述的临床研究的同时,对导致TPMT活性水平表型差异的分子机制的研究也取得了稳步进展。这种理解源于“经典”分子生物学研究策略的应用。首先对人肾TPMT进行纯化,并进行光亲和标记,得到部分氨基酸序列(Van Loon等人,1992年).这些信息使得成功克隆人类TPMT cDNA成为可能(Honchel等人,1993年).以TPMT cDNA为探针进行的Northern blot分析显示,该酶的mRNA在许多人体组织中广泛表达(Lee等人,1995年),提高了TPMT功能扩展到异种生物(如硫嘌呤药物)的生物转化之外的可能性。这个问题将在硫代嘌呤甲基转移酶:悬而未决的问题.然而,在克隆cDNA的时候,直接的目标是确定调控酶活性水平特征的常见遗传多态性的分子基础。通过对TPMT基因结构的了解,实现这一目标将大大简化。然而,使用人类TPMT cDNA克隆该基因的尝试由于位于18号染色体长臂上的一个经过处理的TPMT假基因的存在而变得复杂。Lee等人,1995年).经过处理的假基因的存在不仅仅是智力上的兴趣,因为重要的是,研究人员使用逆转录-聚合酶链反应(RT-PCR)来研究TPMT,避免由于RNA制剂的DNA污染而无意中放大这种无内含子假基因,从而将其误认为是一种变异的mRNA物种。同样重要的是,用于执行基因组DNA扩增的PCR引物包括至少一个基于内含子的引物,以避免tpmt处理的假基因扩增。随后,由于使用锚定PCR技术使得将活性基因映射到6号染色体成为可能,该基因从6号染色体特有的cosmid文库中克隆出来,从而避免了在18号染色体上处理的假基因(Szumlanski等人,1996年).硫代嘌呤甲基转移酶大约有34个碱基长,由10个外显子组成,其中8个编码蛋白质,并映射到染色体6p22.3 (Szumlanski等人,1996年).
文章描述了硫代嘌呤甲基转移酶基因结构还报告了两个单核苷酸多态性(SNPs)存在于白种人低TPMT活性的最常见变异等位基因中-硫代嘌呤甲基转移酶3 *(无花果。4) (Szumlanski等人,1996年).这两个SNPs,一个位于外显子7,另一个位于外显子10,都改变了编码的氨基酸,即它们是非同义的cSNPs。该研究还表明,这两个SNPs的存在与COS-1细胞瞬时表达期间免疫反应性TPMT蛋白水平的降低有关(Szumlanski等人,1996年).后来有报道称,这种效应主要是由于该等位基因编码的变异TPMT蛋白的降解率增加——可能是通过蛋白质组体介导的机制(Tai等,1997).一系列不同的TPMT等位基因现在已经被描述,几乎所有涉及cDNA开放阅读框(ORF)内的snp,导致错义或无意义的密码子(Krynetski等人,1995年;水獭等人,1997年).然而,一个苏格兰民族党硫代嘌呤甲基转移酶内含子9/外显子10剪接破坏了一个扩展亲缘中分离的剪接位点上的规范序列,酶活性水平降低(水獭等人,1998年).这种多态性导致内含子9内的一个隐式剪接位点的激活,以及TPMT mRNA水平的显著降低(水獭等人,1998年).

选择人类TPMT等位基因。
野生型人类TPMT等位基因(硫代嘌呤甲基转移酶* 1)和变异等位基因硫代嘌呤甲基转移酶3 *,硫代嘌呤甲基转移酶b * 3,硫代嘌呤甲基转移酶c * 3.矩形表示外显子,黑色编码区,白色未翻译区。
如前所述,人口研究表明,东亚人RBC TPMT活性的频率分布并不表现为图中所示的三模态频率分布。1(Jang等人,1996;Park-Hah等人,1996).随后的分子流行病学研究表明,最常见的硫代嘌呤甲基转移酶白种人中的变异等位基因,硫代嘌呤甲基转移酶3 *,要么在亚洲人群中不存在,要么出现的频率非常低(水獭等人,1997年;柯利-杜吉德等人,1998年).在这些群体中,最常见的变异等位基因是硫代嘌呤甲基转移酶c * 3,一个等位基因,只包括外显子10 SNP(图。4).尽管目前已知的大多数改变TPMT活性的变异等位基因是由于ORF内的snp,即使具有“高”TPMT活性特征的受试者在其酶活性水平上也表现出广泛的范围(见图。1),据报道,该方差主要是由遗传的影响造成的(Vuchetich等人,1995年).这些观察结果提出了一种可能性,即除了ORF内的snp之外,其他的分子遗传机制可能参与了TPMT活性水平的调节。对这种机制的研究导致在的5 ' -侧翼区域内发现了功能性多态性硫代嘌呤甲基转移酶.
当人类TPMT基因的结构被报道时,人们注意到它没有位于转录起始位点附近的典型TATA盒序列(Szumlanski等人,1996年).然而,该基因的5 '侧区富含gc,具有一系列潜在的Sp1结合位点(Szumlanski等人,1996年).有系统地进行了这些观察Spire-Vayron de la Moureyre等人(1998,1999)的一系列研究表明,5 '的侧翼区域硫代嘌呤甲基转移酶包括多态可变数串联重复(VNTR),其中17或18碱基对重复元件出现4到8次。在研究的原始白种人样本中,最常见的VNTR等位基因有4或5个重复元素(Spire-Vayron de la Moureyre等人,1998,1999).随后一项更大规模的研究证实了这一点硫代嘌呤甲基转移酶VNTR * 4而且* 5是白种人中最常见的等位基因,重复数从3到9不等(Yan等人,2000).最有趣的是,重复单位的总和(即两个等位基因上重复的数量加在一起)与RBC TPMT活性水平(Spire-Vayron de la Moureyre等人,1999;Yan等人,2000).这些经验观察被报告基因构建的研究所证实和扩展,报告基因表达水平随着重复元件数量的增加而降低(Spire-Vayron de la Moureyre等人,1999).这种最近观察到的调节TPMT活性水平的机制是否具有潜在的临床重要性仍有待确定。
TPMT:悬而未决的问题
尽管在过去的二十年中,我们已经了解了很多关于TPMT的知识,包括临床药物遗传学和参与其调控的分子遗传机制,但仍有许多未解之谜。第一个与这种广泛表达的甲基转移酶可能存在内源性底物的可能性有关。目前支持这种可能性的唯一信息是,有报道称,慢性肾功能衰竭患者的血浆中存在甲基硫代甲基mt的甲基受体底物(Pazmiño等人,1980).该底物是内源性还是外源性尚不清楚。如果存在一种内源性底物(尚待确定),那么确定其甲基化降低对基因上TPMT活性水平较低的受试者的可能后果将是有意义的。
其他悬而未决的问题涉及到可能参与调控TPMT活性水平的其他分子遗传机制,这些机制可能超出了目前已知的水平。例如,在一项大群体研究中,具有RBC TPMT活性水平的样本中,16%的表型在杂合范围内,在杂合范围内没有snp硫代嘌呤甲基转移酶外显子或外显子-内含子剪接处(水獭等人,1997年).因此,除了目前已知的多态VNTR之外,是否存在额外的5 '侧区变异,甚至是功能上重要的基于内含子的基因序列变异,仍然没有答案。最后,在白血病患者长期治疗过程中诱导TPMT活性的机制问题(Lennard等人,1990年)仍有待解决。后两个问题都可能具有临床意义。例如,如果一些患者可以诱导这种途径的硫嘌呤代谢,而其他不能,潜在的治疗意义是显而易见的。总之,现在就下结论说我们目前对TPMT分子生物学的理解水平是完整的还为时过早。
结论
TPMT遗传多态性是药物遗传学对医学潜在影响的一个显著例子。硫嘌呤类药物的使用,这类药物是最早将合理药物设计应用于药物开发的一类药物(Elion 1967),持续增加。这些药物广泛用于治疗肿瘤疾病、自身免疫性疾病和器官移植受者(帕特森和蒂德,1975年;林纳德1992).然而,由于硫嘌呤类药物像许多其他抗肿瘤药物一样,具有相对狭窄的治疗指数,因此这些药物的个体化治疗是至关重要的。此外,硫嘌呤,像大多数异种生物一样,有多种生物转化途径(图。2).然而,在过去的二十年中,人们已经清楚地认识到,导致这些药物代谢、毒性和疗效变化的一个重要因素是基因多态性年代甲基化途径的催化作用(Weinshilboum和Sladek, 1980年).事实上,对人类TPMT的系统研究是以探索药物遗传变异的可能性为明确目标开始的,但这一研究领域花了大约20年的时间才发展到目前的状态——无论是就临床意义还是就多态性的分子理解而言——这对于将药物遗传信息转化为临床实践所需的时间来说,是一个有用的教训。然而,由于越来越多的人认识到药物遗传学对催化生物转化途径的酶(如TPMT)以及可能影响药物反应的其他蛋白质的影响,药物治疗可能部分基于药物遗传学信息进行个体化的可能性迅速增加。未来的一个主要挑战将是寻找方法来加速发现过程和将药物遗传学信息转化为有意义的、具有成本效益的临床现实。
鸣谢
我感谢Luanne Wussow在准备这份手稿时给予的帮助。
脚注
将转载请求发送至:Richard Weinshilboum,医学博士,梅奥医学院/梅奥研究生院/梅奥诊所,罗切斯特,MN 55905。电子邮件:weinshilboum.richard在}{mayo.edu
部分由美国国立卫生研究院RO1拨款GM 28157和GM 35720支持。
- 使用的缩写是:
- 硫代嘌呤甲基转移酶
- thiopurine甲基转移酶
- 加拿大皇家银行
- 红细胞
- 6-TGN
- 6-thioguanine核苷酸
- 聚合酶链反应
- 聚合酶链反应
- 单核苷酸多态性
- 单核苷酸多态性
- 羊痘疮
- 开式阅读架
- VNTR
- 可变数串联重复
- 美国药理学和实验治疗学会





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}